Decay of purine and pyrimidine nucleotides. Decay of purine and pyrimidine bases
21. Decay of purine nucleotides. The formation of uric acid. The breakdown of purine nucleotides can occur in various ways. Free adenine and adenine in the composition of nucleotides are deaminated, turning into hypoxanthine and then into xanthine (2,6-dioxipurine), which, under the action of the enzyme xanthine oxidase, is converted into uric acid. Xanthine is also formed during the deamination of guanine. In humans and primates, uric acid is the end product of P. o. and is excreted in the urine. Mammals, except for primates, secrete allantoin, a product of uric acid oxidation, and bony fish, a product of allantoin hydration, allantoic acid. In amphibians and most fish, it is hydrolyzed to urea and glyoxylate.
To the most important violations of P. o. include excessive formation and accumulation of uric acid, such as gout (Gout) and Lesch-Nyhan syndrome. The latter is based on a hereditary deficiency of the enzyme hypoxanthine phosphatidyltransferase, as a result of which free purines are not reused, but are oxidized into uric acid. In children with the Lesha-Nyhan syndrome, inflammatory and dystrophic changes are noted. caused by the deposition of uric acid crystals in the tissues: the disease is characterized by a delay in mental and physical development. Of the other purine bases found in humans, mention should be made of the metabolic precursors of uric acid: aminopurines - guanine, adenine - and oxypurines - hypoxanthine, xanthine.
At present, three main pathways for the formation of uric acid in the body have been proven: a) from purines released during tissue decay; b) from purines contained in food; c) from synthetically formed purines.
The path of formation of uric acid is fundamentally similar to the first one, with the difference that in this case the purine-containing compounds, the conversion of which gives uric acid, are of an alimentary nature. In this case, the cleavage of the protein from the nucleoprotein begins in the stomach under the action of hydrochloric acid with pepsin and ends in the intestine under the influence of trypsin. The resulting nucleic acids under the influence of enzymes of the pancreas and intestinal juice - ribonuclease and deoxyribonuclease - decompose to mononucleotides. The latter, under the action of nucleotidase and nucleosidases of intestinal juice, are cleaved, respectively, to nucleosides and nitrogenous bases. Those and others, as well as some of the mononucleotides, are absorbed in the intestine.
The third pathway for the formation of uric acid in the body, established with the help of C14 and N15 isotopes, etc., goes through the synthesis of purine derivatives, in which glycine, formic and aspartic acids, glutamine, and carbon dioxide take part.
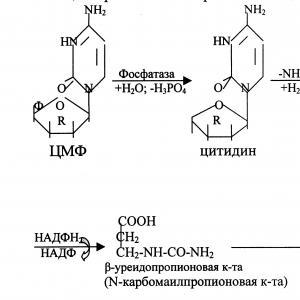
22. Decay of pyrimidine nucleotides. The breakdown of pyrimidine nucleotides begins with the cleavage of a phosphoric acid residue from them, catalyzed by nucleotidases. The resulting nucleosides are further cleaved phosphorolytically to form (deoxy)ribose phosphates and free pyrimidine nitrogenous bases. Cytosine undergoes deamination prior to further degradation. The breakdown of pyrimidine bases is characterized by a reductive pathway followed by opening of the pyrimidine ring. From uracil, the amino acid β-alanine is formed, from thymine - β-aminoisobutyric acid, carbon dioxide and ammonia. Amino acids - degradation products of pyrimidines - can then enter into a variety of metabolic reactions (see Nitrogen metabolism).
Since the intensity of nucleic acid synthesis is regulated at the stage of pyrimidine nucleotide synthesis, P. o. has a significant effect on the exchange of nucleic acids. One of the ways to regulate the synthesis of pyrimidine nucleotides is inhibition by the feedback mechanism: an excess of CTP, the end product of the biosynthetic processes of P. o. allosterically inhibits the enzyme catalyzing the synthesis of carbamoyl aspartate (the first reaction in the biosynthesis of pyrimidines). Pyrimidine nucleotides also inhibit the synthesis of a number of enzymes of pyrimidine metabolism.
In rapidly growing tissues, the activity of enzymes for the breakdown of pyrimidine nucleotides is extremely low; the activity of the enzymes of their synthesis (aspartate carbamoyltransferase, etc.) increases sharply in rapidly dividing tissues, for example, in the liver tissue after partial hepatectomy.
Genetic disorder P. o. may be the cause of hereditary diseases, such as orotaciduria, in which there is excessive excretion in the urine of the degradation product of pyrimidine bases - orotic acid. Pernicious anemia is accompanied by significant disorders of pyrimidine metabolism (see Anemia), and the therapeutic effect of vitamin B12 and folic acid (see Vitamins) in anemia is due to the participation of derivatives of these vitamins as coenzymes in the synthesis of pyrimidine bases.
The breakdown of purine nucleotides.
Adenosine and guanosine, which are formed during the hydrolysis of purine nucleotides, undergo enzymatic degradation with the formation of the final product - uric acid, which is excreted in the urine from the body.
The breakdown of pyrimidine nucleotides.
The initial stages of this process are catalyzed by specific enzymes. End products: CO 2 , NH 3 , urea, β-alanine, β-aminoisobutyric acid. β-alanine is used for the synthesis of muscle dipeptides - carnosine and anserine or is excreted in the urine.

Biosynthesis of purine, pyrimidine nucleotides in tissues.
Biosynthesis of purine mononucleotides.
The initial compound of the synthesis is D-ribose-5-phosphate, which is a product of the pentose phosphate cycle and to which the pyrophosphate group of ATP is transferred. The resulting 5-phosphoribosyl-1-pyrophosphate (FRPP) interacts with glutamine, which is a donor of the NH 2 group, resulting in the formation of β-5-phosphoribosyl-amine. This step becomes key in the synthesis of purines. Then a glycine molecule is attached to the free NH2 group of β-5-phosphoribosyl-amine to form a glycine ribonucleotide. After a few more steps, the first purine nucleotide inosine monophosphate (IMP) is formed, from which the remaining nucleoside phosphates are then synthesized.

Biosynthesis of pyrimidine nucleotides
The initial compounds of this process are carbamoyl phosphate and aspartic acid. From them, through a long chain of reactions, uridine monophosphate (UMP) and the rest of the pyrimidine nucleotides are formed.

2.4. Diseases associated with impaired nucleotide metabolism: gout, Lesch-Nychen syndrome.
Hyperuricemia is an increase in the concentration of uric acid in the blood plasma. Hyperuricemia can lead to gout.
Gout is a disease caused by a disorder in the metabolism of nucleic acids. Uric acid and urate crystals are deposited in cartilage, tendons, articular bags, sometimes in the kidneys, skin, muscles. Around these deposits, inflammation and a granulation shaft are formed that surround the dead tissue, while gouty nodes are formed - tophi (in the joints of the fingers, toes, in the cartilage of the auricle), which is accompanied by deformation and soreness of the affected joints. The characteristic signs of gout include recurring attacks of acute inflammation of the joints (most often small) - acute gouty arthritis. Usually patients are prone to atherosclerosis and hypertension. In their blood there is a high concentration of uric acid - hyperuricemia. Within a few days before an attack of gout, the excretion of water and sodium chloride in the urine increases, i.e. the water-salt balance shifts. As a result, the concentration of uric acid in the blood and its deposition in the tissues increases. Typically, gout is genetically determined and runs in families. It is caused by abnormalities in the work of phosphoribosyl diphosphate (FRDP) synthetase or hypoxanthine guanine or adenine phosphoribosyl transferases. Other characteristic manifestations include nephropathy, in which the formation of urate stones in the urinary tract is observed.
Lesch-Nychen syndrome is a severe form of hyperuricemia that is inherited as an X-linked recessive trait. Appears only in boys. In addition to the symptoms of gout, cerebral palsy, intellectual impairment, attempts to inflict wounds on oneself (bites of lips, fingers) are observed. The disease is associated with a defect in the enzyme hypoxanthine-guanine-phosphoribosyltransferase, which catalyzes the conversion of hypoxanthine and guanine to guanine monophosphate (GMP), so they are converted to uric acid. In the first months of life, neurological disorders are not detected, but pink spots are noted on the diapers, caused by the presence of uric acid crystals in the urine. If left untreated, patients die before the age of 10 due to impaired renal function.
The main drug for the treatment of hyperuricemia is allopurinol (a structural analogue of hypoxanthine).
The breakdown of purine nucleotides.
Adenosine and guanosine, which are formed during the hydrolysis of purine nucleotides, undergo enzymatic degradation with the formation of the final product - uric acid, which is excreted in the urine from the body.
The breakdown of pyrimidine nucleotides.
The initial stages of this process are catalyzed by specific enzymes. Final products: CO2, NH3, urea, β-alanine, β-aminoisobutyric acid. β-alanine is used for the synthesis of muscle dipeptides - carnosine and anserine or is excreted in the urine.
21. Coenzymes: Most enzymes for the manifestation of enzymatic activity require low molecular weight organic compounds of non-protein nature (coenzymes) and / or metal ions (cofactors). Term. "coenzyme" was introduced at the beginning of the 20th century and denoted the part of some enzymes that is easily separated from the protein molecule of the enzyme and removed through a semipermeable membrane during dialysis. Somewhat later, it was found that most enzymes consist of a thermolabile protein part and a thermostable non-protein factor - coenzyme. The protein part is called "apoenzyme", which in the absence of coenzyme does not have catalytic activity. A coenzyme with a protein molecule (apoenzyme) form a holoenzyme molecule with catalytic activity.
Cofactors
More than 25% of all enzymes require metal ions to display full catalytic activity. Let us consider the role of cofactors in enzymatic catalysis.
1. Role of metals in substrate attachment
at the active site of the enzyme
Metal ions perform the function of stabilizers of the substrate molecule, the active center of the enzyme and the conformation of the protein molecule of the enzyme, namely the tertiary and quaternary structures.
Metal ions - stabilizers of the substrate molecule
For some enzymes, the substrate is a complex of the converted substance with a metal ion. For example, for most kinases, one of the substrates is not an ATP molecule, but a Mg2+-ATP complex. In this case, the Mg2+ ion does not interact directly with the enzyme, but participates in the stabilization of the ATP molecule and the neutralization of the negative charge of the substrate, which facilitates its attachment to the active site of the enzyme.

Schematically, the role of a cofactor in the interaction of an enzyme and a substrate can be represented as an E-S-Me complex, where E is an enzyme, S is a substrate, and Me is a metal ion.
An example is the location of substrates in the active site of hexokinase
Hexokinase catalyzes the transfer of the terminal, γ-phosphate residue of the ATP molecule to glucose with the formation of glucose-6-phosphate:

Participation of magnesium ions in the addition of a substrate to the active center of hexokinase. The active center of hexokinase has binding sites for the glucose molecule and the Mg2+-ATP complex. As a result of the enzymatic reaction, the terminal, γ-phosphate residue of the ATP molecule is transferred to glucose with the formation of glucose-6-phosphate.
The Mg2+ ion is involved in the attachment and "correct" orientation of the ATP molecule in the active center of the enzyme, weakening the phosphoester bond and facilitating the transfer of phosphate to glucose.
Metal ions - enzyme active site stabilizers
In some cases, metal ions serve as a "bridge" between the enzyme and the substrate. They act as active site stabilizers, facilitating the attachment of a substrate to it and the occurrence of a chemical reaction. In some cases, the metal ion can contribute to the addition of the coenzyme. The functions listed above are performed by such metals as Mg2+, Mn2+, Zn2+, Co2+, Mo2+. In the absence of metal, these enzymes are inactive. Such enzymes are called "metalloenzymes". Schematically, this process of interaction of the enzyme, substrate, and metal can be represented as follows:
Metalloenzymes include, for example, the pyruvate kinase enzyme (Fig. 2-4), which catalyzes the reaction:

Digestive Enzymes:
2. Stomach
3.Small intestine
Proteases:
Carboxypeptidase
Steapsin, which breaks down fats.
Enzymes of the small intestine
22. Polyenzymatic system: Each cell in the body has its own specific set of enzymes. Some of them are found in all cells, others are present only in some. In a cell, the work of each enzyme, as a rule, is not individual, but is closely related to other enzymes, i.e. polyenzymatic systems, or conveyors, are formed from individual enzymes. The substrate sometimes undergoes a long chain of reactions during its transformation, in which many enzymes participate. The product of the reaction catalyzed by the first enzyme serves as a substrate for the second enzyme, and so on. An example is the process of glycolysis. All glycolysis enzymes are available in a soluble state. A number of enzymes are involved in the conversion of glucose to lactic acid. The position of each enzyme in the chain is determined by its affinity with substrates (beginning with glucose), each of which, respectively, is the product of the reaction catalyzed by the previous enzyme. This increases the rate of enzymatic reactions, and intermediate products do not accumulate in such a chain.
Many polyenzymatic ensembles are structurally associated with any organelle (mitochondria, ribosomes, nucleus) or biomembranes and constitute highly organized systems that provide vital functions, for example, tissue respiration, i.e. transfer of electrons and protons from substrates to oxygen through a system of respiratory enzymes attached to the inner membrane of mitochondria. Some enzymes involved in the reaction of one chain of metabolism are combined into multi-enzyme complexes with a specific function. A typical example of such supramolecular complexes is the pyruvate dehydrogenase complex, which consists of several enzymes involved in the oxidation of pyruvic acid to acetyl-CoA, or fatty acid synthetase, which consists of seven structurally related enzymes that perform the function of fatty acid synthesis.
23. Digestion in metabolism: Metabolism (from the Greek μεταβολή - "transformation, change"), or metabolism - a set of chemical reactions that occur in a living organism to maintain life. These processes allow organisms to grow and reproduce, maintain their structures, and respond to environmental stimuli.
Digestion: Macromolecules such as starch, cellulose, or proteins must be broken down into smaller units before they can be used by cells. Several classes of enzymes are involved in degradation: proteases, which break down proteins into peptides and amino acids, glycosidases, which break down polysaccharides into oligo- and monosaccharides.
Microorganisms secrete hydrolytic enzymes into the space around them, which is different from animals, which secrete such enzymes only from specialized glandular cells. Amino acids and monosaccharides, formed as a result of the activity of extracellular enzymes, then enter the cells using active transport
Digestive Enzymes: Digestive enzymes, digestive enzymes - enzymes that break down complex food components into simpler substances, which are then absorbed into the body. In a broader sense, digestive enzymes also refer to all enzymes that break down large (usually polymeric) molecules into monomers or smaller parts. Digestive enzymes are found in the digestive system of humans and animals. In addition, intracellular enzymes of lysosomes can be attributed to such enzymes. The main sites of action of digestive enzymes in humans and animals are the oral cavity, stomach, and small intestine. These enzymes are produced by glands such as the salivary glands, stomach glands, pancreas, and glands in the small intestine. Part of the enzymatic functions is performed by the obligate intestinal microflora. According to substrate specificity, digestive enzymes are divided into several main groups:
proteases (peptidases) break down proteins into short peptides or amino acids
lipases break down lipids into fatty acids and glycerol
carbohydrases hydrolyze carbohydrates such as starch or sugars into simple sugars such as glucose
Nucleases break down nucleic acids into nucleotides.
1. Oral cavity - The salivary glands secrete alpha-amylase (ptyalin) into the oral cavity, which breaks down high-molecular starch into shorter fragments and into individual soluble sugars (dextrins, maltose, maltriose).
2. Stomach
Enzymes secreted by the stomach are called gastric enzymes.
Pepsin is the main gastric enzyme. Breaks down proteins into peptides.
Gelatinase breaks down gelatin and collagen, the main proteoglycans in meat.
3.Small intestine
pancreatic enzymes
The pancreas is the main gland in the digestive system. It secretes enzymes into the duodenal lumen.
Proteases:
Trypsin is a protease similar to gastric pepsin.
Chymotrypsin is also a protease that breaks down food proteins.
Carboxypeptidase
Several different elastases that break down elastin and some other proteins.
Nucleases that cleave DNA and RNA nucleic acids.
Steapsin, which breaks down fats.
Amylase, which breaks down starch and glycogen, as well as other carbohydrates.
Pancreatic lipase is an essential enzyme in the digestion of fats. It acts on fats (triglycerides) previously emulsified by bile secreted into the intestinal lumen by the liver.
Enzymes of the small intestine
Several peptidases, including:
enteropeptidase - converts trypsinogen into trypsin;
alanine aminopeptidase - cleaves peptides formed from proteins after the action of gastric and pancreatic proteases.
Enzymes that break down disaccharides into monosaccharides:
sucrase breaks down sucrose into glucose and fructose;
maltase breaks down maltose to glucose;
isomaltase breaks down maltose and isomaltose to glucose;
lactase breaks down lactose into glucose and galactose.
Intestinal lipase breaks down fatty acids.
Erepsin, an enzyme that breaks down proteins.
24. Tissue respiration. Cellular or tissue respiration is a set of biochemical reactions occurring in the cells of living organisms, during which carbohydrates, lipids and amino acids are oxidized to carbon dioxide and water. The released energy is stored in the chemical bonds of macroergic compounds (ATP, etc.) and can be used as needed. Included in the group of catabolism processes. On the physiological processes of transporting oxygen to the cells of multicellular organisms and removing carbon dioxide from them, see the article Respiration.
For the first time the essence of breathing was explained by A.-L. Lavoisier (1743-1794), who drew attention to the similarities between the combustion of organic substances outside the body and the respiration of animals. Gradually, the fundamental differences between these two processes became clear: in the body, oxidation proceeds at a relatively low temperature in the presence of water, and its rate is regulated by metabolism. Currently, biological oxidation is defined as a set of reactions of substrate oxidation in living cells, the main function of which is the energy supply of metabolism. In the development of the concepts of biological oxidation in the XX century. the most important contribution was made by A.N. Bach, O. Warburg, G. Kreps, W.A. Engelhardt, V.I. Palladin, V.A. Belitzer, S.E. Severin, V.P. Skulachev.
BIOLOGICAL OXIDATION- a set of enzymatic redox reactions occurring in living cells. In the process of biological oxidation, the breakdown of nutrients occurs, and the energy released in this case is stored in a form convenient for use by cells, the so-called. energy-rich compounds - adenosine triphosphates, etc. These compounds are then used to ensure all vital processes; some of the energy is dissipated as heat. A significant part of biological oxidation reactions is carried out in mitochondria
Anaerobic oxidation of ammonium, anammox - biochemical process of ammonium ion oxidation by nitrite anion under anaerobic conditions. Serves as an energy source for carbon dioxide fixation. Described in the following bacterial genera: Brocadia, Kuenenia, Anammoxoglobus, Jettenia, Scalindua. All of them belong to planctomycetes.
The process was opened in 1986. Now a new technology has been created for the purification of wastewater from nitrogen compounds with the help of bacteria that carry out anaerobic oxidation of ammonium. In Rotterdam (Netherlands), the first treatment plant based on it was built and launched. Important advantages of this technology are the reduction of CO2 emissions into the atmosphere by 85-90% compared to traditional methods, as well as relative cheapness.
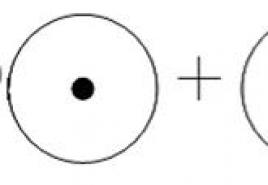
General reaction equation for anaerobic ammonium oxidation:
NH4+ + NO2− → N2 + 2H2O.
Anaerobic oxidation of methane- the process of methane oxidation to carbon dioxide, produced by non-culturable (VBNC) archaea of the ANME-1, ANME-2 and ANME-3 groups, close to Methanosarcinales, in association with sulfate-reducing and denitrifying bacteria in the absence of molecular oxygen in the medium. The biochemistry and prevalence of the process in nature have not yet been studied enough.
26. Pyruvate formed in the reactions of glycolysis (in the cytoplasm) must be transported to the mitochondria. Transport is carried out by a special "shuttle" system. In the mitochondrial matrix, attached to its inner membrane, there is a complex polyenzymatic complex - pyruvate dehydrogenase.
Pyruvate dehydrogenase consists of 60 polypeptide chains, which can be divided into 3 main enzymes: E1 - pyruvate dehydrogenase itself (consists of 24 subunits); E2, dihydrolipoyltransacetylase (also 24 subunits); E3 - dihydrolipoyl dehydrogenase (12 subunits).
The sequence of reactions is shown in Figure 5.12. E1 catalyzes the decarboxylation of PVC with the participation of the coenzyme thiamine pyrophosphate (TPP). The resulting reaction product (hydroxyethyl derivative of TPP) with the participation of E2 reacts with oxidized lipoic acid (LA). Lipoic acid - a low molecular weight nitrogen-containing compound - is a coenzyme E2.
CH2 CH - (CH2) 4 - COOH
Lipoic acid
The disulfide group of LA is capable of being reduced and acetylated. In the reaction catalyzed by dihydrolipoyl transacetylase (E2), acetyl lipoic acid is formed. Further, this compound reacts with coenzyme A (CoA-SH is not its own coenzyme E2) - this forms a reduced form of LA (dihydrolipoic acid) and acetyl-CoA.
Finally, E3 begins to function, the coenzyme of which is FAD: the coenzyme oxidizes dihydrolipoic acid and is itself reduced at the same time (FADH2). The reduced flavin coenzyme reacts with mitochondrial NAD+, in turn reducing it (NADH H+).
Thus, three enzymes that make up a single pyruvate dehydrogenase complex and 5 coenzymes are actually involved in the oxidative decarboxylation of PVA: TPP, LA, and FAD are the complex’s own coenzymes; CoA-SH and NAD+ are external, coming “from outside.” The resulting acetyl-CoA is then oxidized in the Krebs cycle, and hydrogen with NADH H+ enters the mitochondrial respiratory chain.

The mechanism of functioning of the pyruvate dehydrogenase complex
Pyruvate dehydrogenase is distinguished by a large negative redox potential, which is able to provide not only the reduction of NAD +, but also promote the formation of a high-energy thioether bond in acetyl-CoA (CH3-CO ~ ScoA).
With insufficient content in the diet of vitamins that are part of pyruvate dehydrogenase, primarily thiamine, the activity of the enzyme decreases. This leads to the accumulation of pyruvate and lactate in the blood and tissues and the development of metabolic acidosis. With a pronounced deficiency of thiamine, uncompensated acidosis develops, which, if left untreated, leads to death.
^ Regulation of pyruvate dehydrogenase activity
The pyruvate dehydrogenase complex can exist in active and inactive forms. The transition from one form to another is carried out by reversible phosphorylation with the participation of kinase and dephosphorylation with the participation of phosphatase. The phosphorylated form is inactive, while the dephosphorylated form is active.
At a low insulin concentration and a high level of cell energy supply (ATP, acetyl-CoA and NADH H+), this complex is in an inactive state. Activation of the pyruvate dehydrogenase complex is induced by insulin, CoA-SH, pyruvate, ADP, and magnesium ions.
28. Tissue respiration and biological oxidation. The breakdown of organic compounds in living tissues, accompanied by the consumption of molecular oxygen and leading to the release of carbon dioxide and water and the formation of biological forms of energy, is called tissue respiration. Tissue respiration is represented as the final stage of the transformation of monosaccharides (mainly glucose) to the indicated end products, which at different stages includes other sugars and their derivatives, as well as intermediate products of the breakdown of lipids (fatty acids), proteins (amino acids) and nucleic bases. The final reaction of tissue respiration will look like this:
С6Н12О6 + 6O2 = 6СO2 + 6Н2O + 2780 kJ/mol. (one)
Oxygen consumption by tissues depends on the intensity of tissue respiration reactions. The highest rate of tissue respiration is characterized by the kidneys, brain, liver, the lowest - the skin, muscle tissue (at rest). Equation (2) describes the overall result of a multi-step process leading to the formation of lactic acid (see Chapter 10) and proceeding without the participation of oxygen:
C6H12Ob \u003d 2C3H6O3 + 65 kJ / mol. (2)
This path apparently reflects the energy supply of the simplest forms of life that functioned in anoxic conditions. Modern anaerobic microorganisms (carrying out lactic acid, alcohol and acetic acid fermentation) receive energy for life produced in the process of glycolysis or its modifications.
The use of oxygen by cells opens up opportunities for more complete oxidation of substrates. Under aerobic conditions, oxygen-free oxidation products become substrates of the tricarboxylic acid cycle (see Chapter 10), during which the reduced respiratory transporters NADPH, NADH and flavin coenzymes are formed. The ability of NAD+ and NADP+ to play the role of an intermediate hydrogen carrier is associated with the presence of nicotinic acid amide in their structure. When these cofactors interact with hydrogen atoms, reversible hydrogenation (addition of hydrogen atoms) takes place:


In this case, 2 electrons and one proton are included in the NAD+ (NADP+) molecule, while the second proton remains in the medium.
In flavin coenzymes (FAD or FMN), the active part of the molecules of which is the isoalloxazine ring, as a result of reduction, the addition of 2 protons and 2 electrons at the same time is most often observed:


Reduced forms of these cofactors are able to transport hydrogen and electrons to the respiratory chain of mitochondria or other energy-coupling membranes.

Uric acid in humans and a number of animals (primates, birds and some reptiles) is the end product of the breakdown of purine bases and is excreted from the body. The formation of uric acid occurs mainly in the liver. Uric acid is the main breakdown product of nucleotides in humans. The body daily produces 0.5-1 g of uric acid, which is excreted through the kidneys. The blood of a healthy person contains 3-7 mg/dL of uric acid. A chronic increase in the concentration of uric acid (hyperuricemia) often leads to the development of gout - the deposition of poorly soluble uric acid (and its urate salts) in the form of crystals in the blood and tissues. This disease is hereditary and is associated with a defect in the enzyme that catalyzes the reaction of the conversion of hypoxanthine and guanine to inosic acid - IMP (see section 12.3 "Biosynthesis of nucleotides") and GMP, respectively. As a result, hypoxanthine and guanine are not reused for nucleotide synthesis, but are completely converted into uric acid, which leads to hyperuricemia.
Most animals and plants have enzymes that cause further breakdown of uric acid to urea (1) and glyoxalic acid (2):

β-isobutyric acid
H 2 N-COOH → NH 3 + CO 2.
As a rule, the decay products of nucleic acids are excreted from the body. Nucleosides are predominantly absorbed, and in this form, part of the nitrogenous bases can be used for the synthesis of nucleic acids of the body. If the breakdown of nucleosides to free bases occurs, then guanine is not used for synthetic purposes, and the rest in small quantities can participate in the synthesis of nucleic acids.
Biosynthesis of nucleotides
The synthesis of nucleic acids is determined by the rate of synthesis of mononucleotides, while the synthesis of the latter depends on the presence of all three of their components. Pentoses are products of glucose metabolism, phosphoric acid is supplied in sufficient quantities with food. The limiting factor is the biosynthesis of nitrogenous bases.
Related information:
- Handle acid and base solutions with care. If solutions come into contact with skin, contact your teacher immediately.
B. Spare pathways for the synthesis of pyrimidine nucleotides
The use of pyrimidine bases and nucleosides in reutilization reactions prevents the catabolism of these compounds to end products with cleavage of the pyrimidine ring. Some enzymes of nucleotide catabolism are involved in the resynthesis of pyrimidines. Thus, uridine phosphorylase in a reversible reaction can ribosylate uracil to form uridine.
Uracil + Ribose-1-phosphate → Uridine + H 3 RO 4.
The conversion of nucleosides to nucleotides is catalyzed by uridine cytidine kinase.
A part of CMP can be converted into UMP by the action of cytidine deaminase and replenish the reserves of uridyl nucleotides.
CMF + H 2 O → UMF + NH 3.
B. Regulation of the synthesis of pyrimidine nucleotides
The regulatory enzyme in the synthesis of pyrimidine nucleotides is a polyfunctional CAD enzyme. UMP and purine nucleotides allosterically inhibit, and FRDP activates its carbamoyl synthetase activity, while the activity of the aspartate transcarbamoylase domain inhibits CTP but activates ATP (Fig. 10-15).
This method of regulation makes it possible to prevent excessive synthesis not only of UMP, but also of all other pyrimidine nucleotides and to ensure a balanced formation of all four main purine and pyrimidine nucleotides necessary for RNA synthesis.
Orotaciduria
This is the only violation of the synthesis of pyrimidines de novo. It is caused by a decrease in the activity of UMP synthase, which catalyzes the formation and decarboxylation of OMF. Since in embryogenesis from the formation of pyrimidines de novo depends on the provision of DNA synthesis by substrates, then the life of the fetus is impossible in the complete absence of the activity of this enzyme. Indeed, all patients with orotaciduria have marked, albeit very low, activity of UMF synthase. It has been established that the content of orotic acid in the urine of patients (1 g/day or more) significantly exceeds the amount of orotate that is normally synthesized daily (about 600 mg/day). The decrease in the synthesis of pyrimidine nucleotides, observed in this pathology, disrupts the regulation of the CAD enzyme by the mechanism of retroinhibition, which causes hyperproduction of orotate.
Clinically, the most characteristic consequence of orotaciduria is megaloblastic anemia, caused by the inability of the body to provide a normal rate of division of erythrocyte cells. It is diagnosed in children on the basis that it does not respond to treatment with folic acid preparations.
The insufficiency of the synthesis of pyrimidine nucleotides affects the intellectual development, motor ability and is accompanied by disorders of the heart and gastrointestinal tract. The formation of the immune system is disrupted, and there is an increased sensitivity to various infections.
Hyperexcretion of orotic acid is accompanied by disorders of the urinary system and the formation of stones. If left untreated, patients usually die in the first years of life. At the same time, orotic acid does not have a toxic effect. Numerous disturbances in the work of various body systems are caused by "pyrimidine hunger".
To treat this disease, uridine is used (from 0.5 to 1 g / day), which turns into UMF along the "backup" path.
Uridine + ATP → UMF + ADP.
Loading with uridine eliminates "pyrimidine hunger", and since all other nucleotides of the pyrimidine series can be synthesized from UMF, the release of orotic acid decreases due to the restoration of the mechanism of retroinhibition of the CAD enzyme. For patients with orotaciduria, treatment with uridine continues throughout life, and this nucleoside becomes an indispensable nutritional factor for them.
In addition to genetically determined causes, orotaciduria can be observed:
with hyperammonemia caused by a defect in any of the enzymes of the ornithine cycle,
with the exception of carbamoyl phosphate synthetase I. In this case, carbamoyl phosphate synthesized in mitochondria enters the cytosol of cells and begins to be used for the formation of pyrimidine nucleotides. The concentration of all metabolites, including orotic acid, increases. The most significant excretion of orotate is observed with insufficiency of ornithinecarbamoyltransferase (the second enzyme of the ornithine cycle);
in the treatment of gout with allopurinol, which is converted to oxypurinol mononucleotide and becomes a strong inhibitor of UMF synthase. This leads to the accumulation of orotic acid in tissues and blood.
3. Insulin structure, synthesis and secretion. Regulation of insulin synthesis and secretion. Mechanism of action of insulin. The role of insulin and contrainsular hormones (adrenaline and glucagon) in the regulation of metabolism. Changes in hormonal status and metabolism in diabetes mellitus. diabetic coma.
Insulin is a polypeptide consisting of two polypeptide chains. Chain A contains 21 amino acid residues, chain B - 30 amino acid residues. Both chains are interconnected by two disulfide bridges (Fig. 1). Insulin can exist in several forms: monomer, dimer and hexamer. The hexameric structure of insulin is stabilized by zinc ions, which are bound by His residues at position 10 of the B chain of all 6 subunits.
The insulin molecule also contains an intramolecular disulfide bridge connecting the sixth and eleventh residues in the A chain. The insulins of some animals have a significant similarity in primary structure with human insulin.
In both chains, substitutions occur in many positions that do not affect the biological activity of the hormone. Most often, these substitutions are found in positions 8, 9, and 10 of the A chain.
At the same time, substitutions in the positions of disulfide bonds, hydrophobic amino acid residues in the C-terminal regions of the B-chain and C- and N-terminal residues of the A-chain are very rare, which indicates the importance of these regions for the manifestation of the biological activity of insulin. The use of chemical modifications and amino acid substitutions in these regions made it possible to establish the structure of the active center of insulin, which is formed with the participation of the phenylalanine residues of the B chain at positions 24 and 25 and the N- and C-terminal residues of the A chain.
biosynthesis of insulin includes the formation of two inactive precursors, preproinsulin and proinsulin, which, as a result of sequential proteolysis, are converted into an active hormone. The biosynthesis of preproinsulin begins with the formation of a signal peptide on polyribosomes associated with the ER. The signal peptide penetrates the ER lumen and directs the entry of the growing polypeptide chain into the ER lumen. After preproinsulin synthesis is completed, the signal peptide, which includes 24 amino acid residues, is cleaved off (Fig. 2).
Fig.1. The structure of human insulin. A. Primary structure of insulin. B. Model of the tertiary structure of insulin (monomer): 1 - A-chain; 2 - B-chain; 3 - receptor binding site.
Proinsulin (86 amino acid residues) enters the Golgi apparatus, where, under the action of specific proteases, it is cleaved in several sites to form insulin (51 amino acid residues) and a C-peptide consisting of 31 amino acid residues.
Insulin and C-peptide are incorporated into secretory granules in equimolar amounts. In granules, insulin combines with zinc to form dimers and hexamers. Mature granules fuse with the plasma membrane and insulin and C-peptide are secreted into the extracellular fluid by exocytosis. After secretion into the blood, insulin oligomers break down. T 1/2 of insulin in blood plasma is 3-10 minutes, C-peptide - about 30 minutes. The destruction of insulin occurs under the action of the enzyme insulinase mainly in the liver and to a lesser extent in the kidneys.
Regulation of insulin synthesis and secretion. Glucose is the main regulator of insulin secretion, and β-cells are the most important glucose-sensitive cells in the body. Glucose regulates the expression of the insulin gene, as well as the genes of other proteins involved in the metabolism of the main energy carriers. The effect of glucose on the rate of gene expression can be direct, when glucose directly interacts with transcription factors, or secondary, through its effect on the secretion of insulin and glucagon. When stimulated with glucose, insulin is rapidly released from secretory granules, which is accompanied by activation of insulin mRNA transcription.

Rice. 2. Scheme of insulin biosynthesis in β cells of the islets of Langerhans. ER - endoplasmic reticulum. 1 - signal peptide formation; 2 - synthesis of preproinsulin; 3 - signal peptide cleavage; 4 - transport of proinsulin to the Golgi apparatus; 5 - conversion of proinsulin to insulin and C-peptide and incorporation of insulin and C-peptide into secretory granules; 6 - secretion of insulin and C-peptide.
Synthesis and secretion of insulin are not strictly coupled processes. The synthesis of the hormone is stimulated by glucose, and its secretion is a Ca 2+ -dependent process, and in case of Ca 2+ deficiency it decreases even in conditions of high glucose concentration, which stimulates insulin synthesis.
Glucose consumption by β-cells occurs mainly with the participation of GLUT-1 and GLUT-2, and the concentration of glucose in the cells quickly equalizes with the concentration of glucose in the blood. In β-cells, glucose is converted into glucose-6-phosphate by glucokinase, which has a high K m , as a result of which the rate of its phosphorylation almost linearly depends on the concentration of glucose in the blood. The glucokinase enzyme is one of the most important components of the glucose-sensitive apparatus of β-cells, which, in addition to glucose, probably includes intermediate products of glucose metabolism, the citrate cycle, and, possibly, ATP. Mutations in glucokinase lead to the development of a form of diabetes mellitus.