X-ray structural analysis. X-ray diffraction analysis of crystals and interpretation of diffraction patterns What is X-ray diffraction analysis of substances
The name of the analytical method reflects its content - that is, the analysis of the structure of a substance by exposing it to X-rays. The fundamental foundations of the method are related to the theoretical principles of X-ray diffraction by periodic structures, which was discovered by M. Laue in 1912.
X-rays are electromagnetic in nature. Devices that record X-ray quanta are called X-ray diffractometers. The X-ray machine has a control panel, a number of measuring devices and some auxiliary devices.
The main units of the X-ray unit are (Fig. 20):
- - an X-ray detector (counter) with an appropriate electronic circuit and a recording device;
- - radiation source (X-ray machine with X-ray tube);
- - a goniometric device in which the movement of the sample and the counter relative to the primary X-ray beam is carried out.
Rice. twenty. The main components of the DRON diffractometer: 1 - power supply unit; 2 - power supply; 3 - diffraction stand; 4 - x-ray tube; 5 - goniometer; 6 - goniometric attachment; 7 - detection unit; 8 - control complex; 9 - registration block; 10 - counting complex; 11 - self-recording device; 12 - printing device; 13 - perforator
The detector registers at each moment of time the intensity of the scattered radiation in a narrow angular interval of the radiation beam. In this case, a fixed control counter can be used.
The X-ray source is the X-ray tube (Fig. 21), and the X-ray machine serves as the source of electrical energy for the X-ray tube. In the X-ray tube, the energy of the electric current, carried by electrons accelerating to high speeds, is transformed into the energy of electromagnetic radiation.
The objects of study can be substances of various phase states - solid, liquid, gaseous, crystalline and amorphous. However, more often X-ray diffraction methods are used to study solid substances with a crystalline structure, i.e. such substances that are characterized by an ordered, regular arrangement in space of their constituent atoms, ions or complexes. The main pattern of the structure of crystalline substances, namely, the repeatability of the spatial arrangement of particles in three (two) directions with a certain period, reflects the essence of the structure of a crystalline substance, its symmetry and elemental composition.

Rice. 21.
Each substance has only its inherent crystal structure, which determines the individuality of each mineral species or compound, and determines its crystal-physical properties. Several minerals may have the same composition, for example, pyrite and marcasite (FeS), calcite and aragonite (CaCO 3), but the different relative arrangement of atoms and ions in space leads to the individualization of each mineral species. The crystal structure is characterized by a system of parallel atomic planes, more or less populated by atoms, the distances between these planes are called interplanar (d i), and the population density is characterized by the relative intensity of reflection of x-rays (J i). This allows us to solve the inverse problem - having received d and J qualitatively and quantitatively diagnose the mineral structure.
The interaction of X-rays with a crystal can be considered as their reflection by atomic planes and the interference of the reflected rays. The reflected rays, which are maximum in intensity, are observed at certain angles, which depend on the interplanar distances of the reflecting atomic structure and the wavelengths of the initial X-ray radiation (Fig. 22).
This relationship is expressed by the Wulf-Bragg equation:
where u is the angle (Wulf-Bragg) of maximum reflection of x-rays by the atomic plane; d is the distance between the reflecting planes (interplanar distances); l is an integer (the order of reflection); d is the wavelength of the incident X-rays. This equation allows, knowing the value of l and the experimentally measured angles u, to determine the interplanar distances d.

Rice. 22.
The use of this formula allows, taking into account the spatial orientation of atomic planes (h, k, ?) in minerals of different syngonies, to determine the position of the nodes of the atomic (ionic) lattice, indicating the unit cell parameters (a, b, c), where a, b, c - the distance between nodes in the atomic plane and d - the distance between the planes, in accordance with the formula (for the cubic syngony):

The following methods are used to obtain radiographs:
- - Laue method (fixed crystal irradiated with non-monochromatic radiation);
- - crystal rotation method;
- - powder diffraction method (irradiation of a compressed powder with monochromatic radiation).
When studying the crystal structure of a substance by the Laue method, a diffraction pattern of a single crystal is obtained in white (broad spectrum) X-ray radiation. A single crystal is placed under a stream of X-rays, the rays are reflected from atomic planes and fall on an X-ray film (Fig. 23). Scattered rays give point reflections on the film, each of which has its own wavelength l from the polychromatic spectrum. The symmetry in the location of the spots reflects the symmetry of the crystal (Fig. 24).

Rice. 23. Scheme for obtaining a Lauegram (a); view of the diffraction pattern for a crystal (b): ellipses drawn through reflections intersect at a point corresponding to the 4th order symmetry axis (hppt://s-d-p.narod.ru)

Rice. 24.
Ellipses can be drawn through reflections, the point of intersection of which is the axis of symmetry. A diffraction pattern from a single crystal can be obtained by rotating it around an axis perpendicular to the direction of the incident monochromatic beam and parallel to the crystallographic axis, which, as a rule, has small indices.
The diffraction pattern will have a simple form only when the axis of rotation is parallel to some nodal row of the grating. If the film is rolled up in the form of a cylinder, the axis of which coincides with the axis of rotation of the crystal, and the beam is directed perpendicular to this axis (Fig. 25, a), then the planes parallel to the axis of rotation will give a diffraction pattern in the form of points located along a straight line passing through the center film and called the zero layer line of the first kind. Planes oriented obliquely with respect to the axis of rotation will give reflections that form layered lines above and below zero (Fig. 25, b). From the distance between layered lines of the first kind, one can calculate the shortest distance between atoms located along the crystallographic direction parallel to the axis of rotation of the crystal.

Rice. 25. Scheme of X-ray survey according to the rotation method (hppt://bestreferat.ru): 1 - primary beam; 2 - sample (rotates in the direction of the arrow); 3 - cylindrical film; b - typical radiograph of rotation
The crystal structure of a substance can also be determined from powder diffraction patterns obtained from polycrystalline objects. This method of X-ray diffraction study of minerals is called the Debyegram method. It gives a less complete structural characterization of the mineral, but in the absence of large and good quality single crystals, powder methods are very useful. For research by this method, a fine powder of crushed crystals is taken, from which a pressed column is made, or pressed plates. The foundations of this method are related to the assumption that a polycrystalline object contains many differently oriented crystals and it is necessary to create conditions for orienting the largest possible part of them in a position that satisfies the Wolfe-Bragg equation, i.e. obtain maximum angles and reflection intensities (Fig. 26, a). A snapshot of reflected rays is called a debyegram (Fig. 26, b). The analysis of the results is reduced to comparing the Debyeogram of an unknown mineral with the reference images of the standards.

Rice. 26. Scheme of X-ray photography by the powder method (hppt://roman.by): 1 - primary beam; 2 - powder or polycrystalline sample; 3 - film rolled around the circumference; 4 - diffraction cones; 5 - "arcs" on the film, arising when its surface intersects with diffraction cones; b - typical powder x-ray (debyegram)
The above methods of X-ray photography are characterized by the registration of diffracted X-rays on photographic film. In devices called diffractometers, the rays are recorded by counters with which an electronic recording device is connected. The result of the study of the substance on the diffractometer is a diffractogram (Fig. 27), in which the horizontal position of the peaks indicates the magnitude of the angle, and their height characterizes the intensity. Diffractometers of the DRON series are produced in Russia.
X-ray diffraction analysis, performed on advanced equipment and using high-quality reference material to identify the parameters of the crystal lattice, allows:
- - determine the mineral species;
- - determine the mineral variety; (type of crystal lattice);
- - identify structural varieties (subtypes);
- - establish the presence of structural typomorphic features;
- - establish and quantify impurity elements;
- - to reveal the degree of orderliness of the structure and its perfection.
It is a method for studying the structural structure of substances. It is based on the diffraction of an X-ray beam on special three-dimensional crystal lattices. In the study, they use which is approximately 1A, which corresponds to the size of an atom. It must be said that X-ray diffraction analysis, together with neutron and electron diffraction, belongs to diffraction methods for determining the structure of a substance under study.
It helps to explore the atomic structure, space groups, its size and shape, as well as the symmetry group of crystals. Using this technique, metals and their various alloys, organic and inorganic compounds, minerals, amorphous materials, liquids, and gases are studied. In some cases, X-ray diffraction analysis of proteins, nucleic acids and other substances is used.
This analysis helps to establish atomic materials that have a well-defined structure and are natural for x-rays. It should be noted that in the study of other substances, X-ray diffraction analysis requires the presence of crystals, which is an important but rather difficult task.

Discovered by Laue, the theoretical foundations developed by Wolfe and Bragg. Debye and Scherrer suggested using the discovered regularities in the role of analysis. It must be said that at present, X-ray diffraction analysis remains one of the most common methods for determining the structure of substances, since it is simple to perform and does not require significant material costs.
It allows you to explore different classes of substances, and the value of the information obtained determines the introduction of new techniques. So, at first they began to study using the function of interatomic vectors, later direct methods for determining the crystal structure were developed. It is worth noting that the first substances that were studied using X-rays were sodium and potassium chlorides.

The study of the spatial began in the 30s of the last century in Great Britain. The data obtained gave rise to molecular biology, which made it possible to reveal important physicochemical properties of proteins, as well as to create the first model of DNA.
Since the 1950s, computer methods for assembling information that was obtained from X-ray structural analysis began to actively develop.
Today, synchrotrons are used. They are monochrome sources that are used to irradiate crystals. These devices are most effective when using the method of multiwave anomalous dispersion. It should be noted that they are used only in state scientific centers. Laboratories use a less powerful technique, which serves only to check the quality of crystals, as well as to obtain a rough analysis of substances.
Currently, X-ray phase analysis (radiography, or X-ray diffraction) is the most common of the diffraction methods of analysis. It should be noted that diffraction methods are used to study the structure of not only solid crystalline substances, but also liquids and glasses. Liquids and glasses, in which there is a certain fluctuating statistical ordering of structural elements, are also characterized by uneven scattering. In this case, the number and sharpness of the maxima increase as the substance passes into the crystalline state.
Radiography is based on obtaining and analyzing the diffraction pattern resulting from the interference of X-rays scattered by electrons of the atoms of the irradiated object.
The phenomenon of interference of X-rays scattered by a crystal leads to the same results as the specular reflection of rays from the atomic planes of the crystal (Fig. 4.5.
Rice. 4.5. Reflection of x-rays
from the atomic planes of the crystal:
q is the slip angle (Bragg angle);
a is the angle of incidence; d 1 , d 2 - interplanar distances
Reflected rays propagate in a single phase (intensity increases) if the Wulf-Bragg equation is observed:
n l = 2 d sinq,
where n− order of reflection; l is the wavelength of the X-ray beam; d is the distance between the atomic planes of the crystal; q is the grazing angle of the beam.
When the grazing angle changes, when the Wulf-Bragg equation is not observed, the reflected rays propagate in different phases and cancel each other out.
It is obvious that the intensity maxima of the reflected rays will be observed at different values of the angle q for a family of flat grids with different values d. Each crystalline substance has an individual set of families of flat grids, which results in the individuality of the diffraction pattern, i.e., the distribution of reflection intensities depending on the value of the angle q. Therefore, the recording of the diffraction pattern is carried out in the coordinates I− q (intensity of reflected beams − glancing angle).
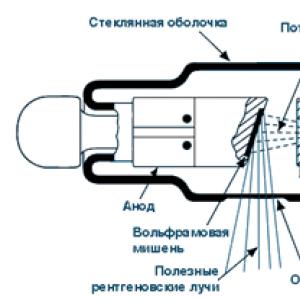
To obtain an X-ray beam, X-ray tubes are used (Fig. 4.6), in which X-rays arise as a result of deceleration of electrons on a metal anode. A stream of electrons emitted by a tungsten filament and accelerated in a voltage field of 30 kV bombards a metal target - the anode of an X-ray tube (made of copper, cobalt or iron). The energy of primary electrons is sufficient to knock out a 1−S electron ( K− copper sheath, fig. 4.7).
Rice. 4.6. X-ray tube diagram:
1 - anode; 2 - tungsten filament; 3 - window
from Ni foil; 4 - x-ray beam
Rice. 4.7. Occurrence of CuK a -radiation
Electrons from outer orbitals immediately move to the vacant seat, i.e., to the inner 1s level. The energy released in this process is emitted in the form of X-rays. The energy of such a transition is a strictly fixed value.
For copper, two types of transitions are possible: 2р ® 1s (K a - radiation; l = 1.5418 Å) and 3р ® 1s (K b - radiation; l = 1.3922 Å). Transitions of the first type occur much more often, so K a radiation is more intense. For the primary x-ray beam, it is desirable to filter out rays with other wavelengths, leaving only the K a radiation. For these purposes, Ni-foil is used, which delays K b radiation.
For an iron anode, K a -radiation corresponds to a wavelength of 0.1936 nm.
There are three classical methods for obtaining a diffraction effect from a crystal:
1) polychromatic method (Laue method), based on the use of a continuous spectrum of x-ray radiation;
2) the rotating single crystal method based on the use of monochromatic radiation;
3) powder method (Debye-Scherrer method), in which the conditions for diffraction of monochromatic X-rays are determined by a large number of differently oriented systems of planes.
It should be noted that in methods 1 and 2 it is necessary to use a single-crystal sample of the test substance. Since, in reality, substances with a polycrystalline structure are most often obtained, method 3 becomes especially important from a practical point of view.
To record the diffraction pattern and the diffraction angle in the powder method, several types of shooting are used; at present, the most commonly used diffractometers are the DRON brand, the general scheme of which is shown in Fig. 4.8.
Rice. 4.8. Diffractometer scheme:
1 - x-ray tube; 2 - diaphragm;
3 - sample; 4 - goniometer; 5 - counter;
6 - circle of movement of the counter
The sample is located in the center of a circle of constant radius along which the counter moves. In this case, the sample rotates simultaneously with the counter. The angular velocity of the counter is twice the angular velocity of the sample. Thus, if the sample is rotated through some angle q, then the counter rotation angle is 2q. X-ray radiation, reflected from the sample, enters the counter, where it is converted into an electrical signal (in the Geiger-Muller counter, the ability of X-rays to ionize gas is used). The X-ray pattern is recorded in coordinates I − 2q. As an example, below is an x-ray of low-temperature quartz (Fig. 4.9).
Rice. 4.9. X-ray diffraction pattern of low-temperature quartz
According to the nature of the tasks to be solved, there are two types of radiographic analysis:
− X-ray diffraction analysis (XRD), designed to determine the parameters and qualitative characteristics of the crystal lattice of the analyzed substance;
− X-ray phase analysis (XRF), which consists in determining the existence of phases (qualitative analysis) and their relative content in the analyzed sample (quantitative analysis).
X-ray diffraction analysis. When studying the structure of a crystalline substance, the following problems arise:
- determination of the size and shape of the elementary cell of the crystal lattice, and, consequently, the number of atoms per cell;
– determination of the specific position (coordinate) of each symmetrically independent cell atom;
− determination of constants of thermal vibrations of atoms and distribution of electron density over atoms and between them.
X-ray diffraction analysis is one of the most informative methods for studying crystalline substances.
X-ray phase analysis. Most materials consist of several phases. Deciphering the qualitative phase composition and the quantitative ratio of different phases, determining the type and state of solid solutions, their possible limiting concentration are the most common materials science problems of X-ray phase analysis.
In general, X-ray phase analysis is based on two assumptions:
– each phase gives a set of diffraction lines inherent only to it (regardless of the presence of other phases);
− the line intensity is proportional to the phase content.
A characteristic of the analysis is its sensitivity - the minimum amount of a substance at which the strongest (reference) line is still noticeable. In general, the XPA sensitivity does not exceed a few percent; for example, for clinker minerals it is 2–3%.
The x-ray diffraction pattern of a multiphase system is the result of superposition of the x-ray diffraction patterns of the individual phases. If the content of the phase is low, then it will be represented by only a limited number of the most intense lines.
Interpretation of radiographs consists in determining the values of interplanar distances d by diffraction peaks and relative intensity of the latter I.
To calculate the values of interplanar distances, the exact value of the angles (2q) for the diffraction peaks (according to their maximum) is set, and the value of the interplanar distance is determined from the corresponding tables d. Then, the correspondence of a set of reflexes that are close in values is compared. d And I reference. The reliability of the identification of the crystalline phase is the higher, the more reflections corresponding to it in the X-ray diffraction pattern. Usually, one can say with certainty about the presence of one or another phase in the presence of at least three reflections corresponding to it.
The search and identification of phases is carried out using the ASTM - ICPDS file cabinet using a PDF powder diffraction file. At present, the ICDD computer database is also widely used for X-ray phase analysis.
X-ray quantitative phase analysis is based on comparing the intensity of the lines of the determined phases with each other or with the intensity of the line of the reference sample obtained on the X-ray pattern by the method of mixing the reference or the method of independent reference.
In this case, in the case of quantitative analysis, the most accurate measurements of the line intensity are required, especially for a phase whose amount is small.
X-ray structural analysis methods for studying the structure of matter by distribution in space and intensities of X-ray radiation scattered on the analyzed object. R. s. but. along with neutron diffraction (See Neutron diffraction) and electron diffraction (See Electron diffraction) is a diffraction structural method; it is based on the interaction of X-rays with the electrons of matter, which results in X-ray diffraction. The diffraction pattern depends on the wavelength of the X-rays used (See X-rays) and the structure of the object. To study the atomic structure, radiation with a wavelength of X-ray structural analysis of 1 Å, i.e., of the order of the size of atoms, is used. R.'s methods with. but. study metals, alloys, minerals, inorganic and organic compounds, polymers, amorphous materials, liquids and gases, protein molecules, nucleic acids, etc. Most successfully R. with. but. used to establish the atomic structure of crystalline bodies. This is due to the fact that Crystals have a strict periodicity in their structure and represent a diffraction grating for X-rays created by nature itself. History reference. The diffraction of X-rays by crystals was discovered in 1912 by the German physicists M. Laue, W. Friedrich, and P. Knipping. Directing a narrow beam of X-rays at a stationary crystal, they registered a diffraction pattern on a photographic plate placed behind the crystal, which consisted of a large number of regularly arranged spots. Each spot is a trace of a diffraction beam scattered by a crystal. radiograph ,
obtained by this method is called the Lauegram (See Lauegram) ( rice. one
). The theory of X-ray diffraction on crystals developed by Laue made it possible to relate the wavelength λ of radiation, the parameters of the unit cell of the crystal a, b, c(see Crystal lattice) ,
angles of the incident (α 0 , β 0 , γ 0) and diffraction (α, β, γ) beams by the ratios: a(cosα - cosα 0) = hλ ,
b(cosβ - cosβ 0) = kλ, (1) c(cosγ - cosγ 0) = lλ ,
In the 50s. R.'s methods of page began to develop rapidly. but. with the use of computers in the technique of the experiment and in the processing of x-ray diffraction information. Experimental methods R. with. but. X-ray cameras and X-ray diffractometers are used to create conditions for diffraction and registration of radiation. The scattered X-ray radiation in them is recorded on photographic film or measured by nuclear radiation detectors. Depending on the state of the sample being studied and its properties, as well as on the nature and amount of information that must be obtained, various methods of R. s are used. but. Single crystals selected for the study of the atomic structure must have dimensions X-ray structural analysis 0.1 mm and, if possible, have a perfect structure. The study of defects in relatively large, almost perfect crystals is carried out by X-ray topography, which is sometimes referred to as x-ray topography. but. The Laue method is the simplest method for obtaining X-ray patterns from single crystals. The crystal in Laue's experiment is stationary, and the X-rays used have a continuous spectrum. Location of diffraction spots on the Laue patterns ( rice. one
) depends on the symmetry of the crystal and its orientation with respect to the incident beam. The Laue method makes it possible to establish whether a crystal under study belongs to one and 11 Laue symmetry groups and to orient it (i.e., determine the direction of the crystallographic axes) with an accuracy of several arc minutes. By the nature of the spots on the Lauegrams, and especially by the appearance of Asterism a, one can reveal internal stresses and some other defects in the crystal structure. The Laue method checks the quality of single crystals when choosing a sample for its more complete structural study. Sample rocking and rotation methods are used to determine the repeat periods (lattice constant) along the crystallographic direction in a single crystal. They allow, in particular, to set parameters but, b, c unit cell of a crystal. This method uses monochromatic X-ray radiation, the sample is brought into oscillatory or rotational motion around an axis coinciding with the crystallographic direction, along which the repeat period is examined. The spots on the rocking and rotation radiographs obtained in cylindrical cassettes are located on a family of parallel lines. The distances between these lines, the radiation wavelength, and the diameter of the X-ray camera cassette make it possible to calculate the required repetition period in the crystal. The Laue conditions for diffraction rays in this method are satisfied by changing the angles included in relations (1) during rocking or rotation of the sample. X-ray methods. For a complete study of the structure of a single crystal by X-ray methods. but. it is necessary not only to establish the position, but also to measure the intensities of as many diffraction reflections as possible, which can be obtained from the crystal at a given radiation wavelength and all possible orientations of the sample. To do this, the diffraction pattern is recorded on photographic film in an X-ray goniometer (See X-ray goniometer) and measured using a Microphotometer a
the degree of blackening of each spot on the x-ray. In an X-ray diffractometer, one can directly measure the intensity of diffraction reflections using proportional, scintillation, and other X-ray photon counters. To have a complete set of reflections, X-ray goniometers take a series of X-ray patterns. On each of them, diffraction reflections are recorded, on the Miller indices of which certain restrictions are imposed (for example, reflections of the type hk 0, hk 1
etc.). Most often, an X-ray goniometric experiment is performed using the Weisenberg methods. Burger ( rice. 2
) and de Jong-Bowman. The same information can be obtained with the help of rocking radiographs. To establish an atomic structure of medium complexity (X-ray structural analysis of 50-100 atoms in a unit cell), it is necessary to measure the intensities of several hundreds and even thousands of diffraction reflections. This very time-consuming and painstaking work is performed by automatic microdensitometers and computer-controlled diffractometers, sometimes for several weeks or even months (for example, in the analysis of protein structures, when the number of reflections increases to hundreds of thousands). By using several counters in the diffractometer, which can register reflections in parallel, the time of the experiment can be significantly reduced. Diffractometric measurements are superior to photographic recording in terms of sensitivity and accuracy. Method for the study of polycrystals (Debye - Scherrer method). Metals, alloys, crystalline powders consist of many small single crystals of a given substance. For their study, monochromatic radiation is used. The X-ray pattern (Debyegram) of polycrystals consists of several concentric rings, each of which merges reflections from a certain system of planes of differently oriented single crystals. Debyegrams of various substances have an individual character and are widely used to identify compounds (including those in mixtures). R.s.a. polycrystals allows you to determine the phase composition of the samples, determine the size and preferred orientation (texturing) of grains in the substance, control the stresses in the sample and solve other technical problems. Study of amorphous materials and partially ordered objects. A clear X-ray pattern with sharp diffraction maxima can only be obtained with a complete three-dimensional periodicity of the sample. The lower the degree of ordering of the atomic structure of the material, the more blurred, diffuse character is the X-ray radiation scattered by it. The diameter of a diffuse ring in an X-ray diffraction pattern of an amorphous substance can serve as a rough estimate of the average interatomic distances in it. With an increase in the degree of order (see Long-Range Order and Short-Range Order) in the structure of objects, the diffraction pattern becomes more complicated and, consequently, contains more structural information. The small-angle scattering method makes it possible to study the spatial inhomogeneities of a substance, the dimensions of which exceed the interatomic distances, i.e. range from 5-10 Å to X-ray structural analysis 10,000 Å. The scattered X-ray radiation in this case is concentrated near the primary beam - in the region of small scattering angles. Small-angle scattering is used to study porous and finely dispersed materials, alloys and complex biological objects: viruses, cell membranes, chromosomes. For isolated protein molecules and nucleic acids, the method allows determining their shape, size, molecular weight; in viruses - the nature of the mutual stacking of their components: protein, nucleic acids, lipids; in synthetic polymers - packing of polymer chains; in powders and sorbents - the distribution of particles and pores by size; in alloys - the occurrence and size of phases; in textures (in particular, in liquid crystals) - the form of packing of particles (molecules) into various kinds of supramolecular structures. The X-ray small-angle method is also used in industry to control the processes of manufacturing catalysts, fine coals, etc. Depending on the structure of the object, measurements are made for scattering angles from fractions of a minute to several degrees. Determination of the atomic structure from X-ray diffraction data. Deciphering the atomic structure of a crystal includes: establishing the size and shape of its elementary cell; determination of whether a crystal belongs to one of the 230 Fedorov (discovered by E. S. Fedorov (see Fedorov)) crystal symmetry groups (see Crystal symmetry); obtaining the coordinates of the basic atoms of the structure. The first and partially second problems can be solved by the Laue methods and rocking or rotation of the crystal. It is possible to finally establish the symmetry group and coordinates of the basic atoms of complex structures only with the help of complex analysis and laborious mathematical processing of the intensity values of all diffraction reflections from a given crystal. The ultimate goal of such processing is to calculate the values of the electron density ρ( x, y, z)
at any point of the crystal cell with coordinates x, y, z. The periodicity of the crystal structure allows us to write the electron density in it through the Fourier series :
where V- unit cell volume, Fhkl- Fourier coefficients, which in R. s. but. are called structural amplitudes, i= hkl and is related to the diffraction reflection, which is determined by conditions (1). The purpose of summation (2) is to mathematically assemble the X-ray diffraction reflections to obtain an image of the atomic structure. To produce in this way image synthesis in R. s. but. This is due to the lack of lenses for x-rays in nature (in visible light optics, a converging lens serves for this). Diffraction reflection is a wave process. It is characterized by an amplitude equal to ∣ Fhkl∣, and phase α hkl(by the phase shift of the reflected wave with respect to the incident), through which the structural amplitude is expressed: Fhkl=∣Fhkl∣(cosα hkl +i sinα hkl). The diffraction experiment makes it possible to measure only reflection intensities proportional to ∣ Fhkl∣ 2 , but not their phases. Phase determination is the main problem in deciphering the crystal structure. The definition of phases of structural amplitudes is fundamentally the same for both crystals consisting of atoms and for crystals consisting of molecules. Having determined the coordinates of atoms in a molecular crystalline substance, it is possible to isolate its constituent molecules and establish their size and shape. It is easy to solve the problem, the reverse of the structural interpretation: the calculation of the known atomic structure of the structural amplitudes, and on them - the intensities of diffraction reflections. The trial and error method, historically the first method of deciphering structures, consists in comparing experimentally obtained ∣ Fhkl∣ exp, with values calculated on the basis of the trial model ∣ Fhkl∣ calc. Depending on the value of the divergence factor A fundamentally new way to deciphering the atomic structures of single crystals was opened by the use of the so-called. Paterson functions (functions of interatomic vectors). To construct the Paterson function of some structure consisting of N atoms, we move it parallel to itself so that the first atom hits the fixed origin first. Vectors from the origin to all atoms of the structure (including a vector of zero length to the first atom) will indicate the position N maxima of the function of interatomic vectors, the totality of which is called the image of the structure in the atom 1.
Let's add more to them N maxima, the position of which will indicate N vectors from the second atom placed at the parallel transfer of the structure to the same origin. After doing this procedure with all N atoms ( rice. 3
), we will get N 2 vectors. The function describing their position is the Paterson function. For the Paterson function R(u, υ, ω)
(u, υ, ω - coordinates of points in the space of interatomic vectors), one can obtain the expression: from which it follows that it is determined by the moduli of structural amplitudes, does not depend on their phases, and, therefore, can be calculated directly from the data of a diffraction experiment. Difficulty in interpreting a function R(u, υ, ω) consists in the need to find the coordinates N atoms from N 2 her
maxima, many of which merge due to overlaps that arise when constructing the function of interatomic vectors. The easiest to decrypt R(u, υ, ω) the case when the structure contains one heavy atom and several light ones. The image of such a structure in a heavy atom will differ significantly from other images of it. Among the various methods that make it possible to determine the model of the structure under study by the Paterson function, the most effective were the so-called superposition methods, which made it possible to formalize its analysis and perform it on a computer. Methods of the Paterson function encounter serious difficulties in studying the structures of crystals consisting of identical or similar atoms in atomic number. In this case, the so-called direct methods for determining the phases of structural amplitudes turned out to be more effective. Taking into account the fact that the value of the electron density in a crystal is always positive (or equal to zero), one can obtain a large number of inequalities to which the Fourier coefficients (structural amplitudes) of the function ρ( x, y, z). Using the methods of inequalities, it is relatively easy to analyze structures containing up to 20–40 atoms in the unit cell of a crystal. For more complex structures, methods based on a probabilistic approach to the problem are used: structural amplitudes and their phases are considered as random variables; distribution functions of these random variables are derived from physical representations, which make it possible to estimate, taking into account the experimental values of the moduli of structural amplitudes, the most probable values of the phases. These methods are also implemented on a computer and make it possible to decipher structures containing 100–200 or more atoms in a unit cell of a crystal. So, if the phases of the structural amplitudes are established, then the electron density distribution in the crystal can be calculated from (2), the maxima of this distribution correspond to the position of the atoms in the structure ( rice. 4
). The final refinement of the coordinates of atoms is carried out on a computer Least squares method om
and, depending on the quality of the experiment and the complexity of the structure, makes it possible to obtain them with an accuracy of up to thousandths of an Å (with the help of a modern diffraction experiment, one can also calculate the quantitative characteristics of thermal vibrations of atoms in a crystal, taking into account the anisotropy of these vibrations). R. s. but. makes it possible to establish more subtle characteristics of atomic structures, for example, the distribution of valence electrons in a crystal. However, this complex problem has so far been solved only for the simplest structures. For this purpose, a combination of neutron diffraction and X-ray diffraction studies is very promising: neutron diffraction data on the coordinates of atomic nuclei are compared with the spatial distribution of the electron cloud obtained using X-ray diffraction. but. To solve many physical and chemical problems, X-ray diffraction studies and resonance methods are jointly used. The pinnacle of R.'s achievements. but. - deciphering the three-dimensional structure of proteins, nucleic acids and other macromolecules. Proteins in natural conditions, as a rule, do not form crystals. To achieve a regular arrangement of protein molecules, proteins are crystallized and then their structure is examined. The phases of the structural amplitudes of protein crystals can only be determined as a result of the joint efforts of radiographers and biochemists. To solve this problem, it is necessary to obtain and study crystals of the protein itself, as well as its derivatives with the inclusion of heavy atoms, and the coordinates of the atoms in all these structures must coincide. About numerous applications of methods of R. of page. but. to study various violations of the structure of solids under the influence of various influences, see Art. Radiography of materials. Lit.: Belov N.V., Structural crystallography, Moscow, 1951; Zhdanov G. S., Fundamentals of X-ray diffraction analysis, M. - L., 1940; James R., Optical principles of X-ray diffraction, trans. from English, M., 1950; Boky G. B., Poray-Koshits M. A., X-ray analysis, M., 1964; Poray-Koshits M.A., Practical course of X-ray diffraction analysis, M., 1960: Kitaygorodsky A.I., Theory of structural analysis, M., 1957; Lipeon G., Cochran V., Determination of the structure of crystals, trans. from English, M., 1961; Weinshtein B.K., Structural electron diffraction, M., 1956; Bacon, J., Neutron Diffraction, trans. from English, M., 1957; Burger M., Structure of crystals and vector space, transl. from English, M., 1961; Guinier A., X-ray diffraction of crystals, trans. from French, Moscow, 1961; Woolfson M. M., An introduction to X-ray crystallography, Camb., 1970: Ramachandran G. N., Srinivasan R., Fourier methode in crystallography, N. Y., 1970; Crystallographic computing, ed. F. R. Ahmed, Cph., 1970; Stout G. H., Jensen L. H., X-ray structure determination, N. Y. - L., . V. I. Simonov. Rice. 9. a. Projection onto the ab plane of the function of interatomic vectors of the mineral baotite O 16 Cl]. The lines are drawn through the same intervals of values of the function of interatomic vectors (lines of equal level). b. The projection of the electron density of baotite onto the ab plane, obtained by deciphering the function of interatomic vectors (a). The electron density maxima (clumps of lines of equal level) correspond to the positions of atoms in the structure. in. Image of a model of the atomic structure of baotite. Each Si atom is located inside a tetrahedron formed by four O atoms; Ti and Nb atoms are in octahedra composed of O atoms. SiO 4 tetrahedra and Ti(Nb)O 6 octahedra in the baotite structure are connected as shown in the figure. Part of the unit cell of the crystal corresponding to Fig. a and b are marked with a dashed line. Dotted lines in fig. a and b determine the zero levels of the values of the corresponding functions. Physical Encyclopedia - X-RAY STRUCTURAL ANALYSIS, the study of the atomic structure of a sample of a substance by the pattern of X-ray diffraction on it. Allows you to establish the distribution of the electron density of a substance, which determines the type of atoms and their ... ... Illustrated Encyclopedic Dictionary - (X-ray diffraction analysis), a set of methods for studying the atomic structure of a substance using X-ray diffraction. According to the diffraction pattern, the distribution of the electron density of the substance is established, and according to it the type of atoms and their ... ... encyclopedic Dictionary - (X-ray structural analysis), research method atomic mol. buildings in c, ch. arr. crystals, based on the study of diffraction arising from the interaction. with the test sample of X-ray radiation of a wavelength of approx. 0.1 nm. Use Ch. arr… Chemical Encyclopedia - (see X-RAY STRUCTURAL ANALYSIS, NEUTRONOGRAPHY, ELECTRONOGRAPHY). Physical Encyclopedic Dictionary. Moscow: Soviet Encyclopedia. Editor-in-Chief A. M. Prokhorov. 1983... Physical Encyclopedia Determination of the structure in in and materials, i.e., finding out the location in space of their constituent structural units (molecules, ions, atoms). In the narrow sense, S. a. determination of the geometry of molecules and mol. systems, which are usually described by a set of lengths ... ... Chemical Encyclopedia
Approximately 10 12 different proteins are found in nature, performing a wide variety of functions. These are protein-enzymes that catalyze biochemical processes in a living cell; and carrier proteins, allowing other molecules to pass through nuclear or cell membranes or move between cells throughout the body; and immunoglobular proteins, characterized by high specificity of interaction with antigens, which leads to the activation of signaling pathways that provide the immune response of cells. These are just a few examples of the unique properties of protein molecules. As Francis Crick figuratively put it, proteins are important primarily because they can perform a wide variety of functions, and with extraordinary ease and grace.
With all their structural and functional diversity, all natural proteins are built from 20 amino acids connected in accordance with the protein synthesis code. Depending on the sequence of amino acid residues in the polypeptide chain, a certain stable three-dimensional structure of the protein is formed, which determines its structural and functional properties. For example, each enzyme is characterized by a well-defined conformation of the active center, which provides specific interaction with substrate molecules and performs a catalytic act. Moreover, for the effective formation of the enzyme-substrate complex, not only the geometric correspondence (complementarity) of the enzyme and substrate molecules is of great importance, but also the formation of hydrogen bonds, electrostatic and hydrophobic interactions between the atoms of the active center of the enzyme and the substrate molecule. Thus, any protein molecule is characterized by a unique structure, which determines the uniqueness of its function.
Elucidation of the spatial organization of proteins is one of the main areas of modern biochemistry. In many cases, knowledge of the structure of a protein and its complex with inhibitors is a decisive factor in the development of drugs.
One of the most important experimental methods that allows one to find out with atomic accuracy what the three-dimensional structure of a protein is, i.e. to determine the spatial coordinates of all atoms of the object under study, is X-ray diffraction, or crystallographic, analysis. Knowing the position of each atom, one can calculate interatomic distances, bond angles, rotation angles around bonds, surface charge distribution, and other details of molecular geometry. These data are needed by chemists, biochemists and biologists who study the relationship between structural characteristics and functional properties, as well as specialists involved in the study of the electronic structure of molecules and molecular interactions. The special place of X-ray diffraction analysis among other experimental methods reflects the fact that since the discovery of X-rays in 1901 to the present, work in this area has been awarded 12 Nobel Prizes.
The use of X-ray diffraction analysis for the study of complex biological objects began after 1953, when Max Perutz, an employee of the Cavendish Laboratory at the University of Cambridge, found a way to determine the structure of large molecules, such as myoglobin and hemoglobin. Since then, X-ray diffraction analysis of protein molecules has helped us understand the chemistry of biological reactions. To date, the structures of about 15 thousand proteins and their complexes with biologically important molecules are known.
X-rays are electromagnetic waves with wavelengths ranging from 0.01 to 10 nm. On the short-wave side, they are adjacent to -rays (wavelengths less than 0.1 nm), on the long-wave side - to ultraviolet (wavelengths of about 10–380 nm).
To conduct an x-ray experiment, monochromatic x-ray radiation (ie, a strictly defined wavelength) is necessary. Various filters and monochromators are used for this purpose.
Usually, when a person hears about an X-ray examination, he remembers the X-ray room in the clinic. In fact, X-ray diffraction analysis has nothing to do with medical research. Medical fluoroscopy is based on the difference in the degree of absorption of X-rays by different tissues, and X-ray crystallography is based on the scattering of X-rays by the electrons of atoms. If in medicine we get an x-ray picture of the object under study, then in x-ray crystallography the pictures do not contain any image of anything.
How is an x-ray experiment set up? The circuit diagram is simple (Fig. 1): the object under study is placed in an X-ray beam and the intensity of the radiation scattered in different directions is measured. The easiest way is to place a photographic film in the path of the beam of rays and, by the degree of darkening of the spot after development, judge the intensity of scattering in this direction. Of course, today there are more advanced methods, but now it does not matter. In this case, it is important that we look not at the intensity of the rays that have passed through the object, but at the intensity of the rays that have arisen where they should not have seemed to be.

Rice. 1. Scheme of X-ray experiment
So, at the input we have an unknown object, at the output - a set of intensities of rays scattered in different directions, or a diffraction pattern. Now it is necessary to connect the information obtained in the experiment with the atomic structure of the object under study. We list the main provisions on which the simplest mathematical model of X-ray scattering is built:
1) the X-ray beam is a plane monochromatic electromagnetic wave;
2) under the influence of this electromagnetic wave, each electron begins to move, which can be described by equations for free charges;
3) the moving electron is, in turn, the source of a new scattered spherical electromagnetic wave propagating in all directions;
4) these new waves are summed up and determine the radiation intensity in the direction of interest to us.
Such a model is called kinematic scattering theory. Its main drawback is that the electron is affected not only by the primary beam, but also by scattered waves, and their influence can change the nature of its motion. An attempt to take these corrections into account is made in a more sophisticated dynamic scattering theory, but for practical applications the simpler kinematic scattering theory is, as a rule, quite sufficient.
The method of X-ray diffraction analysis is based on the diffraction of X-rays on a crystal lattice and therefore is applicable only to substances in the crystalline state. This is due to the fact that in order to register the diffraction pattern of scattering, it is necessary to have a sufficient number of scattering electrons. But if the sample consists of a large number of arbitrarily oriented identical molecules (a solution), then the scattering pattern will be determined by some characteristics averaged over all possible orientations and will hardly allow one to obtain detailed information about the atomic structure. Another thing is if a large number of identical molecules are oriented in the same way. Such an opportunity is given to us by crystalline samples.
In simple terms (and without going into complex mathematical formulations), a crystal is such a sample of the substance under study, in which many (~10 12) identical molecules are in the same orientation and their centers form a regular three-dimensional lattice.
The main feature of the structure of each crystal is that it is built from individual atoms or groups of atoms regularly arranged in space. If each repeating structural unit is replaced by a point, or a node, then a three-dimensional crystal lattice will be obtained (Fig. 2). The lattice can be thought of as a system of identical parallelepipeds. Each such parallelepiped is called the "elementary cell of the crystal" and is described by six parameters: the lengths of the edges (a, b, c) and the angles between them (, , ).
One of the main claims to the method of X-ray diffraction analysis from the very beginning of the study of protein structures is that in life proteins are in solution, while in the study we crystallize them. A logical question arises: does the structure of protein molecules undergo fundamental distortions during crystallization? It is generally accepted that strong distortions still do not occur. The arguments in favor of this position are as follows.
First, a number of proteins retain their enzymatic activity in the crystallized state, i.e. the structure does not change so much that the protein becomes "inoperable". Another consideration: in the crystals of biomacromolecules, a significant volume (from 30 to 80%) is occupied by the solvent, i.e. the packing of protein molecules in a crystal is not dense and is unlikely to cause significant distortions. Some distortions in free loops are possible, but the structure of the active center is preserved. Another confirmation: the alternative determination of the structures of some proteins by the method of two-dimensional nuclear magnetic resonance did not give significant discrepancies with the structures deciphered by X-ray methods.
Monochromatic X-ray radiation, passing through the crystal, is scattered mainly on the electron shells of periodically repeating atoms and forms a diffraction pattern, or X-ray pattern (Fig. 3). Therefore, the experimental x-ray data make it possible to judge the features of the arrangement of electrons in elementary crystalline cells. The electron has wave properties, and its position in space is characterized not by exact coordinates, but by the electron density distribution function (r), which gives the time-averaged number of electrons per 1 3 (cubic angstrom). Based on this function, one can judge the arrangement of atoms in elementary cells, since each atom corresponds to a bunch of electron density of a certain size. Thus, when processing the data of the X-ray experiment, two problems must be solved.

Rice. 3. The diffraction pattern contains all the information about the structure of the protein
1. From the X-ray data, obtain a map of the distribution of electron density (r) in the crystal of the object under study. At this stage, a fundamental difficulty arises (which will be discussed below), associated with the impossibility of obtaining from the experiment all the information necessary to reconstruct the structure under study. Various workarounds are used to obtain the missing piece of information. But there is no universal way, and in each case the researcher chooses the most suitable one, based on his experience and intuition.
2. Based on the electron density distribution map (r), determine the positions of atoms in the object under study. To solve this problem, the structure is repeatedly subjected to software processing and manual refinement to achieve the best match with the electron density.
The main steps in determining the structure of a protein
Isolation, purification
Almost all experimental studies of protein structures begin from this stage. Various biochemical methods are used to obtain the desired protein. The sequence of operations for the isolation of proteins usually comes down to grinding the biological material (homogenization), extracting proteins from it, or rather, transferring the proteins to a dissolved state (extraction) and isolating the protein under study from a mixture of other proteins, i.e. purification and production of an individual protein. At this stage, the greatest difficulty lies in the production of a sufficient amount of pure protein for the experiment.
Crystallization
Obtaining crystals suitable for X-ray diffraction analysis is often a laborious and far from trivial process, especially for complex compounds such as proteins and nucleic acids. The presence of a supersaturated solution is a necessary condition for crystallization. Various methods are used to obtain such a solution. One of them consists in the gradual removal of the solvent by conventional evaporation, which leads to an increase in the concentration of the substance in the solution, which at some point becomes supersaturated. Another method is associated with the use of the dependence of solubility on temperature. For example, if the solubility increases with increasing temperature, you can prepare a saturated solution at a higher temperature and then cool it slowly. Due to the decrease in solubility during the cooling process, a supersaturated solution is obtained. The third method is associated with the introduction into the solution of a substance that causes a decrease in solubility. As such substances, either salts or organic solvents are used. In addition, the solubility of proteins and nucleic acids strongly depends on the pH of the solution, which can also be used to obtain supersaturated solutions.
In practice, everything is much more complicated. Until now, there are no universal methods for selecting optimal crystallization conditions. For each specific protein, the researcher looks for these conditions by changing the type of buffer, pH values, temperature, concentration of the protein itself, precipitating salt, etc. In this work, it is important to find such conditions under which a crystal will be obtained, and salt will not fall out. Therefore, the cultivation of biological crystals is not only a scientific direction, but also an art. Sometimes, in order to force the protein to crystallize, it is centrifuged or even sent to zero gravity.
The choice of crystals for X-ray experiment is carried out using a microscope. For this purpose, a polarizing microscope is especially useful, which makes it possible to establish the presence of defects in a crystal with the help of polarized light. Single crystals with a side size of 0.2–0.6 mm are considered optimal. Crystals should be free from defects and, if possible, with a good cut. The presence of defects leads to errors in the experimental measurement of the diffraction pattern and, as a consequence, to the inaccuracy (and often to the impossibility) of deciphering the crystal structure. With an increase in the complexity of the object under study, the requirements for the quality of crystals increase. What protein crystals look like is shown in Fig. 4.
Rice. Fig. 4. Protein crystals: a – crystals of green fluorescent protein zGFP506; b – crystals of the zGFP506 protein mutant with the N66D amino acid substitution
Unfortunately, it is far from always possible to obtain a crystal of the studied protein; therefore, this stage is the main limitation of the X-ray diffraction analysis of proteins.
X-ray experiment, processing resultsAt present, efforts are being made to use a synchrotron accelerator as a source of x-rays. This is a rather expensive building. Laboratory X-ray machines are also used, but synchrotron radiation has significant advantages.
First, it is the power of the beam. There are two pluses here. The first is clear - the time of the experiment is reduced. The second is that biological crystals tend to break down under the action of X-rays. The destruction process takes a certain time, and if the beam is strong, then you can register the desired pattern before the crystal is destroyed.
Secondly, it is an opportunity to get the desired wavelength. X-ray tubes give a powerful beam of only a fixed wavelength (usually around 1.57), while in the experiment it is often necessary to be able to choose the wavelength. This allows you to make a synchrotron.
The processing of the results of the X-ray experiment is based on a powerful mathematical apparatus, which we will not consider here. When a monochromatic X-ray beam is incident on a crystal oriented in a certain way, the scattering occurs in discrete directions determined by the crystal lattice. The diffraction pattern that appears on the detector film (Fig. 3) is a set of spots or reflections. By measuring the intensity of reflections, one can obtain the values of the modules of the so-called. structural factors (complex numbers) that describe the distribution of electron density in the crystal (r). But in order to unambiguously determine (r), it is also necessary to know the corresponding values of the phases of these factors, information about which is not contained in the diffraction pattern. If phases are determined for any crystal, then the calculation of the positions of the atoms of this crystal does not present fundamental difficulties.
Thus, the central problem of the method of X-ray diffraction analysis, called phase problem, lies in the impossibility of obtaining all the data necessary for the calculation directly from the experiment.
There is currently no general solution to the phase problem. Each case requires a special approach. It is important to understand here that new information does not come from nowhere. In order to obtain phase values, we must either make some new assumptions about the structure and features of the object, or conduct new experiments. Below are the main approaches to solving the "phase problem" used in protein crystallography.
Isomorphic substitution
One can try to introduce a certain label into the crystal molecules – one or several heavy atoms (for example, heavy metal ions), which can either be added to the native structure or can replace some of its atoms (Fig. 5).
By isomorphic insertion of heavy atoms is meant that they are attached to each instance of the molecule in the same place, and the structure of the protein molecule does not change. Then, by conducting an additional X-ray experiment with such a modified compound and determining the changes in the intensity of reflections compared to the native protein, one can obtain additional information about the values of the phases. The difficulty of this method lies in the fact that it is not always possible to obtain a good isomorphic derivative, and also in the need for an additional x-ray experiment.
The isomorphic substitution method is the main method for solving the phase problem in determining the structure of biological macromolecules. This method itself arose quite a long time ago, but it was when working with proteins that it acquired an exceptionally important role. There are two reasons for this:
1) for a long time it was the only method to solve the phase problem for proteins;
2) it is for proteins that it is “quite easy” to obtain isomorphic derivatives. The latter is due to the fact that protein crystals are quite friable - in them from 30 to 70% of the volume is occupied by a solvent, i.e. crystals have "voids" where additional atoms can fit.
Using the anomalous scattering effect
This method is based on varying the wavelength of the X-ray radiation incident on the crystal close to the values at which the resonance effect (and the corresponding anomalous scattering) is observed for several "special" atoms contained in the structure of the macromolecule. If there are no abnormally scattering atoms in the protein, sometimes you can try to attach them chemically. Diffraction patterns are obtained for several wavelengths of the incident beam, and based on the analysis of the differences in the intensities of the corresponding reflections, the phase values are estimated.
The success of the anomalous scattering method, as well as isomorphic substitution, largely depends on the possibility of experimentally obtaining derivatives with the required properties.
The two methods mentioned above correspond to an attempt to solve the phase problem with additional information obtained from additional experiments. The following method is used in a situation where we know the structure of a closely related (homologous) protein.
Molecular displacement method
In biology, a situation is common when there are rows of objects that are similar to each other, i.e. having structural homology. Such homology can be exhibited, for example, by proteins of the same type isolated from different organisms. In this case, one can hope that the phases of structural factors calculated using the known atomic model of a homologous protein will be a fairly good initial approximation to the values of the unknown phases corresponding to the object under study. Combining them further with the moduli of structure factors measured in the experiment for the object under study, we can obtain a good approximation to the desired electron density distribution.
However, in order to hope for success along this path, it is necessary, at least to begin with, to “place” a known homologous object in the same place and in the same orientation as the protein under study. The procedure for creating such a “computer hybrid”, in which a molecule of another is placed inside the unit cell of a crystal of one protein, is called the method of molecular substitution. It is possible to judge how close the obtained placement is to reality by comparing the moduli of structural factors calculated by the model with the values obtained in the experiment. Of course, such a substitution is just a speculative procedure, and no chemical substitution occurs.
"Direct" Methods
Unlike previous approaches, these methods do not rely on an additional experiment or information about the structure of a homologous object, but on an almost philosophical idea about the atomicity of the object under study. In crystallography, “direct” methods are understood as strategies for determining structures that use as starting information only a set of reflection intensities obtained in an X-ray experiment. To determine the phases of structural factors, they use a probabilistic approach. "Direct" methods are more objective in the sense that they depend only on the application of mathematical relationships.
On the basis of "direct" methods, the structures of most low-molecular compounds are determined. These methods do not require any additional experiments, or fine biochemical work to obtain isomorphic derivatives, or the presence of known homologous structures, but, unfortunately, are not yet applicable to protein structures due to fundamental limitations on the number of atoms of the structure under study.
If both the modulus and the phase of the structure factors are known, then we can recover the distribution (r) by calculating the inverse Fourier transform. This is not a computationally difficult task from a modern point of view, and this step stands out because it sums up an important stage of work. We finally get the opportunity to "look" at the object of interest to us. And by how “clear” the image turned out, one can judge the success of all previous stages of work. And in case of failure, repeat all over again.
The next step is to build an approximate atomic model based on the calculated electron density distribution maps. This work requires the maximum use of human intelligence and is carried out by qualified specialists.
Using special computer programs, the researcher manually enters the atoms of the protein structure into the electron density map obtained at the previous stage (Fig. 6).