Строение нитрогруппы. Методы получения и химические свойства нитросоединений
Нитрование ароматических соединений – основной путь получения нитросоединений. Процесс нитрования как частный случай электрофильного замещения в ароматическом ряду был уже рассмотрен раньше. Поэтому представляется целесообразным сосредоточить внимание на синтетических возможностях этой реакции.
Достаточно легко и с хорошим результатом нитруется уже сам бензол
В более жестких условиях способен нитроваться и нитробензол с образованием м -динитробензола

Из–за дезактивирующего влияния двух нитрогрупп ввести третью нитрогруппу в м -динитробензол удается лишь с большим трудом. 1,3,5-Три- нитробензол был получен с выходом 45% в результате нитрования м -динитробензола при 100-110 о С и продолжительности реакции 5 суток.
Трудности получения тринитробензола прямым нитрованием бензола привели к разработке косвенных методов. По одному из них более доступный, чем тринитробензол, тринитротолуол окисляется до 2,4,6-тринитробензойной кислоты, которая легко декарбоксилируется при нагревании в воде

Точно также к косвенным методам приходится прибегать и при необходимости получения 1,2-динитробензола. В этом случае обычно используется способность аминогруппы окисляться до нитрогруппы в о -нитроанилине

Даже в тех случаях, когда получение нитросоединений нитрованием не должно было бы встретить особых затруднений, приходится обращаться к косвенным методам. Так, не удается получить пикриновую кислоту нитрованием фенола, т.к. азотной кислотой фенол не нитруется, а окисляется. Поэтому обычно используется следующая схема

Тонкости данной схемы в том, что из-за дезактивации кольца хлором и двумя уже имеющимися нитрогруппами, не удается вводить в него третью нитрогруппу. Поэтому хлор в динитрохлорбензоле предварительно замещается на гидроксил, чему нитрогруппы как раз способствуют (бимолекулярное замещение). Образовавшийся динитрофенол легко принимает еще одну нитрогруппу, не окисляясь в заметной степени. Имеющиеся нитрогруппы предохраняют бензольное кольцо от окисления.
Еще одним неочевидным способом получения пикриновой кислоты является сульфирование фенола до 2,4-фенолдисульфокислоты с последующим нитрованием образовавшегося соединения. При этом одновременно с нитрованием происходит замещение сульфогрупп на нитрогруппы

Один из важнейших ароматических нитропроизводных – тринитротолуол в технике получают нитрованием толуола, которое протекает по следующей схеме

Химические свойства
Ароматические нитросоединения способны реагировать как с участием бензольного кольца, так и нитрогруппы. Указанные структурные элементы влияют на реакционную способность друг друга. Так, под воздействием нитрогруппы нитробензол в реакции электрофильного замещения вступает неохотно и новый заместитель принимает в м -положение. Нитрогруппа влияет не только на реакционную способность бензольного кольца, но и на поведение соседних функциональных групп в химических реакциях.
Рассмотрим реакции ароматических нитросоединений за счет нитрогруппы.
16.2.1. Восстановление. Одной из важнейших реакций нитросоединений является их восстановление до ароматических аминов, широко используемых при производстве красителей, лекарственных препаратов и фотохимикатов.
Возможность преобразования нитрогруппы в аминогруппу восстановлением нитросоединений впервые была показана Зининым в 1842 году на примере реакции нитробензола с сульфидом аммония

В последующем восстановление ароматических нитросоединений было предметом глубокого изучения. При этом установлено, что в общем случае восстановление носит сложный характер, протекает через ряд стадий с образованием промежуточных продуктов. Амины являются лишь конечным продуктом реакции. Результат восстановления определяется силой восстанавливающего агента и рН-среды. При электрохимическом восстановлении состав продуктов зависит от величины потенциала на электродах. Варьируя указанными факторами можно задержать процесс восстановления на промежуточных стадиях. В нейтральной и кислой средах восстановление нитробензола идет последовательно через образование нитрозобензола и фенилгидроксиламина

Когда восстановление проводится в щелочной среде, образовавшийся нитрозобензол и фенилгидроксиламин получают возможность конденсироваться между собой с образованием азоксибензола, в котором атомы азота и кислорода связаны между собой семиполярной связью

Предполагаемый механизм конденсации напоминает механизм альдольной конденсации
Восстановление азоксибензола в анилин идет через азо- и гидразобензолы
Все упомянутые выше промежуточные продукты восстановления нитробензола в анилин могут быть получены либо непосредственно из нитробензола, либо исходя друг из друга. Вот некоторые примеры


16.2.2. Влияние нитрогруппы на реакционную способность других функциональных групп. При изучении ароматических галогенпроизводных мы уже встречались со случаем, когда подходящим образом расположенная нитрогруппа (нитрогруппы) существенно влияла на нуклеофильное замещение галогена (бимолекулярное замещение ароматически связанного галогена). На примере о - и п -динитробензолов было установлено, что нитрогруппа может способствовать нуклеофильному замещению не только галогена, но даже другой нитрогруппы

Механизм бимолекулярного замещения нитрогруппы на гидроксильную группу можно представить как следующий двухстадийный процесс

Карбанион, образующийся на первой стадии рассматриваемой реакции, резонансно стабилизирован из-за вклада предельной структуры 1, в которой нитрогруппа оттягивает электроны именно с того углерода бензольного кольца, у которого их избыток.
Особенностью нуклеофильного замещения одной нитрогруппы под влиянием другой нитрогруппы является то, что реакция весьма чувствительна к расположению нитрогрупп относительно друг друга. Известно, что м -динитробензол не реагирует со спиртовым раствором аммиака даже при 250 о С.
Другими примерами содействия нитрогруппы замещению, в данном случае гидроксила, являются превращения пикриновой кислоты

16.2.3. Комплексообразование с ароматическими углеводородами. Характерным свойством ароматических нитросоединений является их склонность образовать комплексы с ароматическими углеводородами. Связи в таких комплексах носят электростатический характер и возникают между электронодонорными и электроноакцепторными частицами. Рассматриваемые комплексы называют π -комплексами или комплексами с переносом заряда.
π –Комплексы в большинстве случаев представляют собой кристаллические вещества с характерными температурами плавления. При необходимости π -комплекс может быть разрушен с выделением углеводорода. Благодаря сочетанию указанных свойств π -комплексы используются для выделения, очистки и идентификации ароматических углеводородов. Особенно часто для комплексообразования используется пикриновая кислота, комплексы которой неправильно называются пикратами.
Глава 17
Амины
По степени замещения водородных атомов в аммиаке на алкильные и арильные заместители различают первичные, вторичные и третичные амины. В зависимости от природы заместителей амины могут быть жирно – ароматическими и чисто ароматическими.
Ароматические амины называют путем прибавления окончания «амин» к названию групп, связанных с азотом. В сложных случаях аминогруппу с меньшим заместителем обозначают приставкой «амино» (N–метиламино-, N,N-диметиламино), которую прибавляют к названию более сложного заместителя. Ниже приведены наиболее часто встречающиеся амины и их названия

Методы получения
Со многими из методов получения аминов мы уже встречались при изучении алифатических аминов. При приложении этих методов к синтезу ароматических аминов встречаются некоторые особенности, поэтому, не опасаясь повторов, рассмотрим их.
17.1.1. Восстановление нитросоединений. Восстановление нитросоединений – основной метод как лабораторного, так и промышленного получения аминов, который можно осуществить несколькими способами. К ним относятся каталитическое гидрирование, восстановление атомарным водородом и химическое восстановление.
Каталитическое восстановление осуществляется молекулярным водородом в присутствии тонко измелченных никеля или платины, комплексных соединений меди на носителях. При выборе катализатора и условий восстановления надо иметь в виду, что при этом могут восстановиться и другие функциональные группы. Кроме того, каталитическое восстановление нитросоединений должно проводиться с соблюдением определенной осторожности из-за чрезвычайной экзотермичности реакции.
При использовании в качестве химического восстановителя сульфида аммония появляется возможность восстановления только одной из нескольких нитрогрупп

17.1.2. Аминирование галогенпроизводных. Известны трудности, которые возникают при аминировании ароматических галогенпроизводных по механизму «элиминирования – присоединения». Однако, как уже об этом говорилось не раз, электроноакцепторные заместители в бензольном кольце, расположенные надлежащим образом, значительно облегчают замещение галогена в арилгалогенидах, направляя процесс по бимолекулярному механизму. Для сравнения ниже приведены условия аминирования хлорбензола и динитрохлорбензола

17.1.3. Расщепление по Гофману. Расщепление амидов кислот по Гофману позволяет получить первичные амины, которые содержат на один углерод меньше, чем исходные амиды

Реакция протекает с миграцией фенила от карбонильного углерода к атому азота (1,2-фенильный сдвиг) по следующему предполагаемому механизму

17.1.4. Алкилирование и арилирование аминов. Алкилирование первичных и вторичных ароматических аминов галогеналкилами или спиртами позволяет получить вторичные и третичные жирноароматические амины

К сожалению, при участии в реакции первичных аминов получается смесь. Этого можно избежать, если исходный амин предварительно проацилировать, а уже потом проалкилировать

Такой прием защиты аминогруппы позволяет получить чистые вторичные ароматические амины, а также третичные амины с разными замещающими радикалами

Арилирование аминов дает возможность получить чистые вторичные и третичные ароматические амины
Химические свойства
Ароматические амины реагируют как с участием аминогруппы, так и бензольного кольца. При этом каждая функциональная группа испытывает влияние другой группы.
Реакции по аминогруппе
Благодаря наличию аминогруппы ароматические амины вступают в многочисленные реакции. Некоторые из них были уже рассмотрены: алкилирование, ацилирование, реакция с альдегидами с образованием азометинов. Другие реакции, которым будет уделено внимание, легко предсказуемы, однако им присущи определенные особенности.
Основность
Наличие неподеленной пары электронов у атома азота, которые могут быть представлены на образование связи с протоном, обеспечивает ароматическим аминам основные свойства

Интерес представляет сопоставление основности алифатических и ароматических аминов. Как уже было показано при изучении алифатических аминов, об основности аминов удобно судить по константе основности К в

Сравним основности анилина, метиламина и аммиака
Аммиак 1,7 . 10 -5
Метиламин 4,4 . 10 -4
Анилин 7,1 . 10 -10
Из этих данных видно, что появление электронодонорной метильной группы повышает электронную плотность у атома азота и приводит к усилению основности метиламина по сравнению с аммиаком. В то же время фенильная группа более чем в 10 5 раз ослабляет основность анилина по сравнению с аммиаком.
Уменьшение основности анилина по сравнению с алифатическими аминами и аммиаком может быть объяснено сопряжением неподеленной пары электронов азота с секстетом электронов бензольного кольца

Это снижает способность неподеленной пары электронов присоединять протон. Еще более эта тенденция сказывается у ароматических аминов, которые содержат в бензольном кольце электроноакцепторные заместители

Так, м -нитроанилин как основание в 90 раз слабее, чем анилин.
Как и можно было ожидать, электронодонорные заместители в бензольном кольце усиливают основность ароматических аминов

Жирноароматические амины под влиянием алкильной группы проявляют бóльшую основность, чем анилин и амины с электроноакцепторными группами в кольце.
Предельные нитросоединения с открытой цепью (нециклические) имеют общую формулу С n H 2n+1 NO 2 . Они изомерны алкилнитритам (эфирам азотистой кислоты) с общей формулой R-ONO. Различия следующие:
Алкилнитриты имеют более низкие температуры кипения
Нитросоелинения сильно полярны и имеют большой дипольный момент
Алкилнитриты легко омыляются щелочами и минеральными кислотами с образованием соответствующих спиртов и азотистой кислоты или ее соли.
Восстановление нитросоединений приводит к аминам, алкилнитритов - к спиртам и гидроксиламину.
Получение
По реакции Коновалова - нитрованием парафинов разбавленной азотной кислотой при нагревании. В реакцию жидкофазного нитрования вступают все углеводороды, однако скорость реакции невелика и выходы малы. Реакция сопровождается окислением и образованием полиниросоединений. Наилучшие результаты получаются с углеводородами, содержащими третичный атом углерода. Парофазное нитрование протекает при 250-500 о С с парами азотной кислоты. Реакциясопровождается крекингом углеводородов, в результате получаются всевозможные нитропроизводные, и окислением, в результате которого образуются спирты, альдегиды, кетоны, кислоты. Также образуются и непредельные углеводороды. Азотная кислота может быть заменена оксидами азота. Нитрование протекает по механизму S R .
Взаимодействием галогенпроизводных предельных углеводородов с нитритом серебра при нагревании. Атакующей частицей является ион NO 2 - , проявляющий двойственную реакционную способность (амбиденность), т.е. присоединять радикал по азоту (S N 2) с образованием нитросоединения R-NO 2 или кислороду с образованием эфира азотистой кислоты R-O-N=O.(S N 1). Механизм реакции и ее направление сильно зависят от природы растворителя. Сольватирующие растворители (вода, спирты) благоприятствуют образованию эфира.
Химические свойства
При восстановлении нитросоединений образуются первичные амины:
Первичные и вторичные нитросоединения растворимы в щелочах с образованием солей. Водородные атомы при углероде, связанном с нитрогруппой активируются, в результате в щелочной среде ниросоединения перегруппировываются в аци-нитро-форму:

При обработке минеральной кислотой щелочного раствора нитросоединения образуется сильно кислая аци-форма, которая быстро изомеризуется в обычную нейтральную форму:
Нитросоединения относят к псевдокислотам. Псевдокислоты нейтральны и не электропроводны, но тем не менее образуют нейтральные соли щелочных металлов. Нейтрализации нитросоединений щелочами происходит медленно, а истинных кислот - мгновенно.
Первичные и вторичние нитросоединения реагируют с азотистой кислотой, третичные не реагируют:

Щелочные соли нитроловых кислот в растворе имеют красный цвет, псевдонитролы – синий или зеленовато-синий.
Первичные и вторичные ниросоединения конденсируются в присутствии щелочей с альдегидами, образуя нитроспирты (нуклеофильное присоединение):

Аци-формы первичных и вторичных нитросоединений в водных растворах при действии минеральных кислот образуют альдегиды или кетоны:

Первичные нитросоединения при нагревании с 85%-ной серной кислотой переходят в карбоновые кислоты с отщеплением гидроксиламина. Это происходит в результате гидролиза образующейся аци-формы.
Известны также N- и О-нитро-соединения (см. и Нитраты органические).
Нитрогруппа имеет строение, промежуточное между двумя предельными резонансными структурами:
ФИЗИЧЕСКИЕ СВОЙСТВА НЕКОТОРЫХ АЛИФАТИЧЕСКИХ НИТРОСОЕДИНЕНИЙ

* При 25°С. ** При 24°С. *** При 14°С.
В ИК спектрах нитросоединений присутствуют две характеристич. полосы, соответствующие антисимметричным и симметричным валентным колебаниям связи N-О: для первичных нитросоединений соотв. 1560-1548 и 1388-1376 см -1 , для вторичных 1553-1547 и 1364-1356 см -1 , для третичных 1544-1534 и 1354-1344см -1 ; для нитроолефинов RCH=CHNO 2 1529-1511 и 1351-1337 см -1 ; для динитроалканов RCH(NO 2) 2 1585-1575 и 1400-1300 см -1 ; для тринитроалканов RC(NO 2) 3 1610-1590 и 1305-1295 см -1 ; для ароматических Н. 1550-1520 и 1350-1330 см -1 (электроноакцепторные заместители сдвигают высокочастотную полосу в область 1570 -1540, а электронодонорные - в область 1510-1490 см -1); для Н. 1610-1440 и 1285-1135 см -1 ; нитроновые эфиры имеют интенсивную полосу при 1630-1570 см, связь С-N-слабую полосу при 1100-800 см -1 .
В УФ спектрах алифатических нитросоединений l макс 200-210 нм (интенсивная полоса) и 270-280 нм (слабая полоса); для и эфиров нитроновых кислот соотв. 220-230 и 310-320 нм; для гем-динитросоед. 320-380 нм; для ароматических Н. 250-300 нм (интенсивность полосы резко снижается при нарушении копланарности).
В спектре ПМР хим. сдвиги a-Н-атома в зависимости от строения 4-6 м.д. В спектре ЯМР 14 N и 15 N хим. сдвиг 5 от - 50 до + 20 м.д.
В масс-спектрах алифатических нитросоединений (за исключением CH 3 NO 2) пик мол. отсутствует или очень невелик; осн. процесс фрагментации - отщепление NO 2 или двух с образованием фрагмента, эквивалентного . Для ароматических нитросоединений характерно присутствие пика мол. ; осн. пик в спектре соответствует , получаемому при отщеплении NO 2 .
Химические свойства.
Нитрогруппа - одна из наиб. сильных электроноакцепторных групп и способна эффективно делокализовать отрицат. заряд. В ароматич. соед. в результате индукционного и особенно она влияет на распределение : ядро приобретает частичный положит. заряд, который локализован главным образом в орто- и пара-положениях; константы Гаммета для группы NO 2 s м 0,71, s n 0,778, s + n 0,740, s - n 1,25. Т. обр., введение группы NO 2 резко увеличивает реакц. способность орг. соед. по отношению к нуклеоф. реагентам и затрудняет реакции с электроф. реагентами. Это определяет широкое применение нитросоединений в орг. синтезе: группу NO 2 вводят в нужное положение орг. соед., осуществляют разл. реакции, связанные, как правило, с изменением углеродного скелета, и затем трансформируют в др. ф-цию или удаляют. В ароматич. ряду часто используют и более короткую схему: нитрование-трансформация группы NO 2 .
Мн. превращения алифатических нитросоединений проходят с предварит. в нитроновые кислоты или образованием соответствующего . В растворах равновесие обычно практически полностью сдвинуто в сторону С-формы; при 20 °С доля аци-формы для 1 10 -7 , для нитропропана 3 . 10 -3 . Нитроновые кислоты в своб. виде, как правило, неустойчивы; их получают осторожным подкислением Н. В отличие от Н. они проводят ток в растворах и дают красное окрашивание с FeCl 3 . Аци-Н.-более сильные СН-кислоты (рК а ~ 3-5), чем соответствующие нитросоединения (рК а ~ 8-10); кислотность нитросоединений повышается с введением электроноакцепторных заместителей в a-положение к группе NO 2 .
Образование нитроновых кислот в ряду ароматических Н. связано с бензольного кольца в хиноидную форму; например, образует с конц. H 2 SO 4 окрашенный солеобразный продукт ф-лы I, о-нитротолуол проявляет в результате внутримол. переноса с образованием ярко-синего О-производного:

При действии оснований на первичные и вторичные Н. образуются нитросоединений; амбидентные в реакциях с электрофилами способны давать как О-, так и С-производные. Так, при алкилировании Н. алкилгалогенидами, триалкилхлорсиланами или R 3 O + BF - 4 образуются продукты О-алкилирования. Последние м.б. получены также при действии диазометана либо N,О-бис-(триметилсилил)аце-тамида на нитроалканы с рК а

Ациклич. алкиловые эфиры нитроновых кислот термически нестабильны и распадаются по внутримол. механизму:
р-цию можно использовать для получения . Более стабильны силиловые эфиры. Об образовании продуктов С-алкилирования см. ниже.
Для нитросоединений характерны реакции с разрывом связи С-N, по связям N=O, O=N О, C=N -> О и реакции с сохранением группы NO 2 .
Р-ц и и с р а з р ы в о м с в я з и С-N. Первичные и вторичные Н. при нагр. с минер. кислотами в присутствии спиртового или водного раствора образуют карбонильные соед. (см. Нефа реакция). Р-ция проходит через промежут. образование нитроновых кислот:

В качестве исходных соед. можно использовать силиловые нитроновые эфиры. Действие сильных кислот на алифатические нитросоединения может приводить к гидроксамовым кислотам, например:

Метод используют в промышленности для синтеза СН 3 СООН и из нитроэтана. Ароматические нитросоединения инертны к действию сильных кислот.
Алифатические нитросоединения, содержащие подвижный Н в b-положении к группе NO 2 , при действии оснований легко элиминируют ее в виде HNO 2 с образованием . Аналогично протекает термич. разложение нитроалканов при температурах выше 450°. Вицинальные динитросоед. при обработке Са в гексамстаноле отщепляют обе группы NO 2 , Ag-соли непредельных нитросоединений при потере групп NO 2 способны димеризоваться:

Нуклеоф. замещение группы NO 2 не характерно для нитроалканов, однако при действии тиолат-ионов на третичные нитроалканы в апротонных растворителях группа NO 2 замещается на . Р-ция протекает по анион-радикальному механизму. В алифатич. и гетероциклич. соед. группа NO 2 при относительно легко замещается на нуклеофил, например:

В ароматич. соед. нуклеоф. замещение группы NO 2 зависит от ее положения по отношению к др. заместителям: группа NO 2 , находящаяся в мета-положении по отношению к электроноакцепторным заместителям и в орто- и пара-положениях к электронодонорным, обладает низкой реакц. способностью; реакц. способность группы NO 2 , находящейся в орто- и пара-положениях к электроноакцепторным заместителям, заметно увеличивается. В некоторых случаях заместитель вступает в орто-положение к уходящей группе NO 2 (напр., при нагр. ароматических Н. со спиртовым раствором KCN, реакция Рихтера):

Р-ц и и п о с в я з и N = O. Одна из важнейших реакций-вос-становление, приводящее в общем случае к набору продуктов:

Азокси-(II), азо-(III) и гидразосоед. (IV) образуются в щелочной среде в результате промежуточно возникающих нитрозосоед. с и . Проведение процесса в кислой среде исключает образование этих веществ. Нитрозосоед. восстанавливаются быстрее, чем соответствующие нитросоединения, и выделить их из реакц. смеси, как правило, не удается. Алифатические Н. восстанавливаются в азокси- или при действии Na, ароматические - при действии NaBH 4 , обработка последних LiAlH 4 приводит к . Электрохим. ароматических Н. при определенных условиях позволяет получить любое из представленных производных (за исключением нитрозосоед.); этим же методом удобно получать из мононитроалканов и амидоксимы из гем-динитроалканов:
Р-ции по связям O = N О и C = N О. Нитросоединения вступают в реакции 1,3-диполярного , например:

Наиб. легко эта реакция протекает между нитроновыми эфира-ми и или . В продуктах (моно- и бициклич. диалкоксиаминах) под действием нуклеоф. и электроф. реагентов связи N - О легко расщепляются, что приводит к разл. алифатич. и гетеро-циклич. соед.:

В препаративных целях в реакции используют стабильные силиловые нитроновые эфиры.
Р-ц и и с с о х р а н е н и е м г р у п п ы NO 2 . Алифатические Н., содержащие a-Н-атом, легко алкилируются и ацилируются с образованием, как правило, О-производных. Однако взаи-мод. дилитиевых первичных Н. с алкилгалогенидами, ангидридами или галогенангидридами карбоновых кислот приводит к продуктам С-алкилирования или С-ацилирования, например:
Известны примеры внутримол. С-алкилирования, например:
Первичные и вторичные нитросоединения реагируют с алифатич. и СН 2 О с образованием р-аминопроизводных (р-ция Манниха); в реакции можно использовать предварительно полученные метилольные производные нитросоединений или аминосоед.:


Легко вступают в реакции присоединения нитроолефины: с в слабокислой или слабощелочной среде с послед. ретрореакцией Анри они образуют карбонильные соед. и нитроалканы; с нитросоединениями, содержащими a-Н-атом,-поли-нитросоединений; присоединяют и др. СН-кислоты, такие, как , и малоновой кислот, реактивы Гриньяра, а также нуклеофилы типа OR - , NR - 2 и др., например:

Нитроолефины могут выступать в роли диенофилов или диполярофилов в реакциях и циклоприсое-динения, а 1,4-динитродиены-в роли диеновых компонентов, например:

Получение.
В промышленности низшие нитроалканы получают жидкофазным (р-ция Коновалова) или парофазным (метод Хэсса) смеси , и , выделяемых из природного или полученных переработкой (см. Нитрование). Таким методом получают и высшие Н., например нитроциклогексан - полупродукт в произ-ве капролактама.
В лаборатории для получения нитроалканов применяют азотной кислотой соед. с активир. метиленовой группой; удобный метод синтеза первичных нитроалканов -нитрование 1,3-индандиона с послед. щелочным a-нитрокетона:

Алифатические нитросоединения получают также взаимод. AgNO 2 с алкилгалогенидами или NaNO 2 с эфирами a-галогенкарбо-новых кислот (см. Мейера реакция). Алифатические Н. образуются при и ; -способ получения гем-ди- и гем-тринитросоединений, например:

Нитроалканы м.б. получены нагреванием ацилнитратов до 200 °С.
Мн. методы синтеза нитросоединений базируются на олефинов , HNO 3 , нитрония, NO 2 Cl, орг. нитратами и т.п. Как правило, при этом получают смесь виц-динитросоединений, нитронитратов, нитронитритов, непредельных нитросоединений, а также продуктов сопряженного присоединения группы NO 2 и растворителя или продуктов их , например:
3. Нитросоединения. Способы получения и важнейшие свойства.
Нитросоединения - органические вещества, содержащие нитрогруппу -N0 2 .
Общая формула - R-NO 2 .
В зависимости от радикала R, различают алифатические(предельные и непредельные), ациклические, ароматические и гетероциклические нитросоединения. По характеру углеродного атома, с которым связана нитрогруппа, нитросоединения подразделяются на первичные , вторичные и третичные .
Методы получения алифатических нитросоединений
Прямое нитрование алканов в жидкой или газовой фазе под действием 50-70%-ной водной азотной кислоты при 500-700 о С или тетраоксида азота при 300-500 о С имеет промышленное значение только для получения простейших нитроалканов, так как нитрование в этих условиях всегда сопровождается крекингом углеводородов и приводит к сложной смеси самых разнообразных нитросоединений. Эта реакция не получила по этой причине широкого распространения.
Наиболее общим лабораторным методом получения нитроалканов до сих пор остается реакция алкилирования нитрит-иона, открытая В. Мейером еще в 1872 году. В классическом методе В. Мейера нитрит серебра взаимодействует с первичными или вторичными алкилбромидами и алкилиодидами в эфире, петролейном эфире или без растворителя при 0-20 о С с образованием смеси нитроалкана и алкилнитрита.
Нитрит-ион принадлежит к числу вырожденных амбидентных анионов с двумя независимыми нуклеофильными центрами (азот и кислород), которые не связаны в единую мезомерную систему.
Реакционная способность амбидентного нитрит-иона с двумя независимыми нуклеофильными центрами (азот и кислород) резко отличается от реакционной способности енолят-ионов с двумя нуклеофильными центрами, связанными в единую мезомерную систему.
Соотношение продуктов N- и O- алкилирования (нитроалкан / алкилнитрит) в реакции Мейера алкилбромидов и иодидов с нитритом серебра решающим образом зависит от природы алкильной группы в алкилгалогениде. Выходы первичных нитроалканов достигают 75-85%, однако они резко снижаются до 15-18% для вторичных и до 5% для третичных нитроалканов.
Таким образом, ни третичные, ни вторичные алкилгалогениды непригодны для синтеза нитроалканов при взаимодействии с нитритом серебра. Реакция Мейера является по-видимому лучшим способом получения первичных нитроалканов, арилнитрометанов и -нитроэфиров карбоновых кислот.

Для получения нитроалканов следует использовать только алкилбромиды и алкилиодиды, поскольку алкилхлориды, алкилсульфонаты и диалкилсульфаты не взаимодействуют с нитритом серебра. Из,-дибромалканов легко получаются,-динитроалканы.
Н. Корнблюм (1955 г.) предложил модифицированный общий метод получения первичных и вторичных нитроалканов, а также,-динитроалканов и нитрозамещенных кетонов.
Этот метод основан на алкилировании нитритов щелочных металлов первичными или вторичными алкилгалогенидами в диполярном апротонном растворителе ДМФА. Для того чтобы предотвратить последующее нитрозирование нитроалкана параллельно образующимся алкилнитритом, в реакционную смесь необходимо вводить мочевину или многоатомные фенолы - резорцин или флороглюцин. Выход первичных нитроалканов по этому методу не превышает 60 %, т.е. ниже чем при алкилировании нитрита серебра (75-80%). Однако, вторичные нитроалканы можно получать с хорошим выходом алкилированием нитрита натрия в ДМФА.

Третичные алкилгалогениды подвергаются элиминированию под действием нитрит-иона и не образуют нитросоединений. Эфиры -хлор- или -бромзамещенных кислот гладко превращаются в эфиры -нитрозамещенных кислот с выходом 60-80 % при взаимодействии с нитритом натрия в ДМСО или ДМФА.
Другой общий метод синтеза нитроалканов состоит в окислении оксимов кетонов с помощью трифторперуксусной кислоты в ацетонитриле.

Кроме оксимов можно окислять также и первичные амины перуксусной кислотой или м-хлорпербензойной кислотой:

Более ста лет назад Г. Кольбе описал метод получения нитрометана при взаимодействии хлорацетата натрия и нитрита натрия в водном растворе при 80-85 o С:
Промежуточно образующийся анион нитроуксусной кислоты декарбоксилируется до нитрометана. Для получения гомологов нитрометана способ Кольбе не имеет значения из-за низкого выхода нитроалканов. Идея этого метода была остроумно использована при разработке современного общего метода получения нитроалканов. Дианионы карбоновых кислот нитруются под действием алкилнитрата с одновременным декарбоксилированием -нитрозамещенной карбоновой кислоты.

Нитрование карбанионов с помощью алкилнитратов широко используется и для получения, - динитроалканов. С этой целью енолят-ионы циклических кетонов обрабатывают двумя эквивалентами алкилнитрата. Раскрытие цикла с последующим декарбоксилированием приводит к,-нитроалкану.

Методы получения ароматических нитросоединений
Ароматические нитросоединения получают чаще всего нитрованием аренов, что было подробно рассмотрено при изучении электрофильного ароматического замещения. Другой общий метод получения нитроаренов заключается в окислении первичных ароматических аминов с помощью трифторперуксусной кислоты в хлористом метилене. Трифторперуксусную кислоту получают непосредственно в реакционной смеси при взаимодействии ангидрида трифторуксусной кислоты и 90 %-ной перекиси водорода. Окисление аминогруппы до нитрогруппы с помощью трифторперуксусной кислоты имеет значение для синтеза нитросоединений, содержащих в орто- и пара- положениях другие электроноакцепторные группировки, например, для получения орто- и пара- динитробензола, 1,2,4- тринитробензола, 2,6 - дихлорнитробензола и др..

Реакции алифатических нитросоединений:
Первичные и вторичные нитроалканы находятся в таутомерном равновесии с аци-формой нитросоединения, называемой иначе нитроновой кислотой.

Из двух таутомерных форм нитро-форма гораздо более стабильна и преобладает в равновесии. Для нитрометана при 20 о концентрация аци-формы не превышает 110 -7 от доли нитроалкана, для 2-нитропропана она возрастает до 310 -3 . Количество аци-формы возрастает для фенилнитрометана. Изомеризация аци-нитросоединения в нитросоединение происходит медленно. Это дает возможность определить концентрацию аци-формы титрованием бромом с очень высокой степенью точности.
Малая скорость взаимопревращения двух таутомерных форм позволила А.Ганчу еще в 1896 году выделить в индивидуальном виде обе таутомерные формы фенилнитрометана. Фенилнитрометан нацело растворяется в холодном водном растворе гидроксида натрия. При обработке его водной уксусной кислотой при 0 о образуется бесцветное твердое вещество, представляющее собой аци-форму фенилнитрометана. Она мгновенно окрашивается в красный цвет при обработке хлоридом железа (III) и количественно титруется бромом.
При стоянии твердая аци-форма медленно изомеризуется в более стабильную жидкую форму фенилнитрометана. Для простых нитроалканов, например, нитрометана, нитроэтана и 2-нитропропана аци-форму не удается выделить в индивидуальном виде, так как она довольно легко при 0 о изомеризуется в нитро-форму и о содержании аци-формы можно судить только по данным титрометрического бромирования.
Концентрация двух таутомерных форм для любого соединения всегда обратно пропорциональна кислотности таутомерных форм, аци-форма нитроалканов во всех случаях является более сильной кислотой по сравнению с нитро-формой. Для нитрометана в воде рКа ~ 10,2, тогда как для его аци-формы CH 2 =N(OH)-O рКa ~ 3,2 . Для 2-нитропропана это различие значительно меньше, рКа (СH 3) 2 CHNO 2 составляет 7,68, а для (CH 3) 2 C=N(OH)-O рКа равно 5,11.
Различие в величинах рКа для двух форм не является неожиданным, поскольку аци-форма представляет собой О-Н кислоту, тогда как нитро-форма относится к С-Н кислотам. Напомним, что аналогичная закономерность наблюдается для кето- и енольных форм карбонильных и 1,3-дикарбонильных соединений, где енол оказывается более сильной О-Н кислотой по сравнению с С-Н кислотностью кето-формы.
Аци-нитросоединения представляют собой довольно сильные кислоты, образующие соли даже при взаимодействии с карбонатом натрия в отличие от нитро-формы нитроалканов, которая не реагирует с карбонат-ионом. Таутомерные превращения обоих форм нитроалканов катализируются как кислотами, так и основаниями, аналогично енолизации альдегидов и кетонов.
Реакции амбидентных анионов нитроалканов.
При действии основания как на нитро-форму, так и на аци-форму нитросоединения, образуется общий для них обоих мезомерный амбидентный анион, в котором заряд делокализован между атомами кислорода и углерода.

Амбидентные анионы нитроалканов во всех отношениях являются близкими аналогами енолят-ионов карбонильных соединений и для них характерны те же самые реакции замещения, что и для енолят-ионов.
Наиболее типичными и важными реакциями с участием анионов нитроалканов являются: галогенирование, алкилирование, ацилирование, конденсации с карбонильными соединениями, реакции Манниха и Михаэля - все те, которые типичны и для енолят-ионов. В зависимости от природы электрофильного агента и в некоторой степени от строения нитроалкана замещение может происходить с участием либо кислородного, либо углеродного, либо обоих центров амбидентного аниона нитроалкана.
Галогенирование щелочных солей нитросоединений осуществляется только по атому углерода, реакцию можно остановить на стадии введения одного атома галогена.

Нитрозирование первичных нитроалканов также осуществляется только по атому углерода и приводит к образованию так называемых нитроловых кислот.

Вторичные нитроалканы в тех же условиях дают псевдонитролы.

Нитроловые кислоты бесцветны и при встряхивании с раствором гидроксида натрия образуют соли, окрашенные в красный цвет.
В отличие от них псевдонитролы обладают в нейтральной среде голубой окраской. Эти соединения могут быть использованы для идентификации первичных и вторичных нитроалканов. Третичные нитроалканы не взаимодействуют при 0 о или ниже с азотистой кислотой.
Алкилирование амбидентных анионов нитроалканов протекает в отличие от галогенирования и нитрозирования преимущественно по атому кислорода с образованием в качестве промежуточных соединений эфиров аци-формы, которые носят название нитроновых эфиров. Эфиры аци-формы нитроалканов могут быть выделены в индивидуальном виде при алкилировании солей нитроалканов тетрафторборатами триалкилоксония в хлористом метилене при -20 о.
Нитроновые эфиры термически неустойчивы и выше 0-20о подвергаются окислительно-восстановительному распаду на оксим и карбонильное соединение.

Оксим всегда образуется как конечный продукт восстановления нитроалкана, тогда как альдегид оказывается конечным продуктом окисления алкилирующего агента. Эта реакция нашла широкую область применения в синтезе ароматических альдегидов.
При взаимодействии щелочных солей 2-нитропропана с замещенными бензилгалогенидами конечными продуктами являются оксим ацетона и ароматический альдегид.

Еще более важную роль играет алкилирование амбидентных анионов нитроалканов под действием аллил-галогенидов для получения ,-ненасыщенных альдегидов.

Как следует из приведенных выше примеров, в отличие от енолят-ионов анионы нитроалканов подвергаются региоселективному О-алкилированию. Такое резкое различие в поведении двух родственных классов амбидентных анионов обусловлено высокой степенью локализации заряда на атоме кислорода аниона нитроалкана.
При наличии в бензилгалогениде одной или нескольких сильных электроноакцепторных группировок, таких как NO 2 , NR 3 , SO 2 CF 3 и др., происходит изменение механизма реакции и ее региоселективности. В этом случае наблюдается С-алкилирование аниона нитроалкана по механизму с участием анион-радикалов, который по своей сути аналогичен S RN 1 -механизму ароматического нуклеофильного замещения.

Открытие анион-радикального механизма С-алкилирования нитроалканов и других амбидентных анионов позволило Н.Корнблюму в 1970-1975 годах разработать исключительно эффективный метод алкилирования амбидентных анионов с помощью -нитрозамещенных сложных эфиров, нитрилов и др.,способствующих реализации анион-радикального цепного процесса.

Следует отметить, что в этих реакциях замещение происходит даже у третичного атома углерода.
С-Алкилирование удается сделать практически единственным направлением реакции в случае алкилирования дианионов нитроалканов. Дианионы нитроалканов образуются при обработке первичных нитроалканов двумя эквивалентами н-бутиллития в ТГФ при -100 о.
Эти дианионы подвергаются также региоселективному С-ацилированию при взаимодействии с ацилгалогенидами или ангидридами карбоновых кислот.


Конденсация анионов нитроалканов с карбонильными соединениями (реакция Анри).
Конденсация анионов первичных и вторичных нитроалканов с альдегидами и кетонами приводит к образованию -гидроксинитроалканов или продуктов их дегидратации - ,-непредельных нитросоединений.

Эта реакция была открыта Л. Анри в 1895 году и может рассматриваться как разновидность альдольно-кротоновой конденсации карбонильных соединений.
В конденсации принимает участие анион нитроалкана, а не карбонильного соединения, поскольку кислотность нитроалканов (рКа ~ 10) на десять порядков выше кислотности карбонильных соединений (рКа ~ 20).
Эффективными катализаторами реакции Анри являются гидроксиды, алкоксиды и карбонаты щелочных и щелочноземельных металлов.
Щелочность среды следует тщательно контролировать для того чтобы исключить альдольную конденсацию карбонильных соединений или реакцию Каниццаро для ароматических альдегидов. Первичные нитроалканы могут также реагировать с двумя молями карбонильного соединения, поэтому соотношение реагентов следует соблюдать очень тщательно. При конденсации ароматических альдегидов обычно образуются только -нитроалкены и реакцию очень трудно остановить на стадии образования -гидроксинитроалкана.
Присоединение анионов нитроалкана к активированной двойной связи по Михаэлю и реакция Манниха с участием нитроалканов.
Анионы первичных и вторичных нитроалканов присоединяются по кратной связи
,-непредельных карбонильных соединений, сложных эфиров и цианидов аналогично тому, как это происходит при присоединении к активированной двойной связи енолят-ионов.

Для первичных нитроалканов реакция может идти и дальше с участием второго моля CH 2 =CHX. Анионы нитроалканов в реакции присоединения по Михаэлю получают обычным образом с помощью этилата натрия или диэтиламина в качестве основания.
-Нитроалкены также могут быть использованы в качестве акцепторов Михаэля в реакции присоединения стабилизированных сопряжением карбанионов. Присоединение анионов нитроалканов к - нитроалкенам является одним из наиболее простых и удобных методов синтеза алифатических динитросоединений.

Такого типа присоединение может происходить и в условиях реакции Анри в результате дегидратации продукта конденсации альдегида или кетона с нитроалканом и последующего присоединения нитроалкана.

Первичные и вторичные алифатические амины вступают в реакцию Манниха с первичными и вторичными нитроалканами и формальдегидом.
По своему механизму и области применения эта реакция ничем не отличается от классического варианта реакции Манниха с участием карбонильных соединений вместо нитроалканов.
Реакции ароматических нитросоединений:
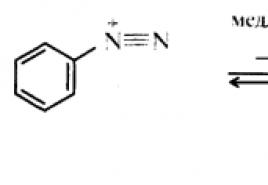
Нитрогруппа отличается высокой стабильностью по отношению к электрофильным реагентам и разнообразным окислителям. Большинство нуклеофильных агентов за исключением литий- и магнийорганических соединений, а также литийалюминийгидрида не действуют на нитрогруппу. Нитрогруппа относится к числу превосходных нуклеофильных групп в процессах активированного ароматического нуклеофильного замещения (S N A r). Так, например, нитрогруппа в 1,2,4-тринитробензоле легко замещается под действием гидроксид-, алкоксид-ионов или аминов.

Наиболее важной реакцией ароматических нитросоединений является восстановление их допервичных аминов.
Эта реакция была открыта в 1842 году Н.Н.Зининым, который впервые восстановил нитробензол до анилина действием сульфида аммония. В настоящее время для восстановления нитрогруппы в аренах до аминогруппы в промышленных условиях применяется каталитическое гидрирование. В качестве катализатора используют медь на силикагеле в качестве носителя. Катализатор готовят нанесением карбоната меди из суспензии в растворе силиката натрия и последующим восстановлением водородом при нагревании. Выход анилина над этим катализатором составляет 98 %.

Иногда в промышленном гидрировании нитробензола до анилина в качестве катализатора используют никель в комбинации с оксидами ванадия и алюминия. Такой катализатор эффективен в интервале 250-300 о и легко регенерируется при окислении воздухом. Выход анилина и других аминов составляет 97-98 %. Восстановление нитросоединений до аминов может сопровождаться гидрированием бензольного кольца. По этой причине для получения ароматических аминов избегают использовать в качестве катализаторов платину. палладий или никель Ренея.
Другим методом восстановления нитросоединений является восстановление металлом в кислой или щелочной среде.
Восстановление нитрогруппы до аминогруппы происходит в несколько стадий, последовательность которых сильно различается в кислой и щелочной среде. Рассмотрим последовательно процессы, протекающие при восстановлении нитросоединений в кислой и щелочной среде.
При восстановлении в кислой среде в качестве восстановителя применяют железо, олово, цинк и соляную кислоту. Эффективным восстановителем нитрогруппы является хлорид олова (II) в соляной кислоте. Этот реагент особенно эффективен в тех случаях, когда в ароматическом нитросоединении есть другие функциональные группы: CHO, COR, COOR и др., чувствительные к действию других восстановителей.
Восстановление нитросоединений до первичных аминов в кислой среде происходит ступенчато и включает три стадии с переносом двух электронов на каждой стадии.

В кислой среде каждый из промежуточных продуктов быстро восстанавливается до конечного продукта анилина и их не удается выделить в индивидуальном виде. Однако, в апротонных растворителях в нейтральной среде можно зафиксировать промежуточные продукты восстановления.
При восстановлении нитробензола натрием или калием в ТГФ сначала образуется анион-радикал нитробензола за счет переноса одного электрона от щелочного металла.

Катион щелочного металла связан в контактную ионную пару с атомом кислорода нитрогруппы анион-радикала. При дальнейшем восстановлении анион-радикал превращается в дианион, который после протонирования дает нитрозобензол.
Нитрозобензол, также как и другие ароматические нитрозосоединения, обладает высоким окислительным потенциалом и очень быстро восстанавливается до N-фенилгидроксиламина. Поэтому нитрозобензол не удается выделить в качестве промежуточного продукта восстановления, хотя данные электрохимического восстановления однозначно указывают на его образование.
Дальнейшее восстановление нитрозосоединений до N-арилгидроксиламина включает две аналогичные стадии одноэлектронного восстановления до анион-радикала и далее до дианиона нитрозосоединения, который при протонировании превращается в N-арилгидроксиламин.
Последняя стадия восстановления арилгидроксиламина до первичного амина сопровождается гетеролитическим расщеплением связи азот-кислород после протонирования субстрата.
В нейтральном водном растворе можно получить фенилгидроксиламин в качестве продукта восстановления нитробензола. Фенилгидроксиламин получается при восстановлении нитробензола цинком в водном растворе хлорида аммония.
Арилгидроксиламины легко восстанавливаются в амины при обработке железом или цинком и соляной кислотой.
Поскольку фенилгидроксиламин является промежуточным продуктом восстановления, его можно не только восстановить до анилина, но и окислить до нитрозобензола.

Это, вероятно, один из лучших методов получения ароматических нитрозосоединений, которые не удается иным способом выделить в качестве промежуточного продукта восстановления нитросоединений.
Ароматические нитрозосоединения легко димеризуются в твердом состоянии, причем их димеры бесцветны. В жидком и газообразном состоянии они мономерны и окрашены в зеленый цвет.

Восстановление нитросоединений металлами в щелочной среде отличается от восстановления в кислой среде. В щелочной среде нитрозобензол быстро взаимодействует со вторым промежуточным продуктом восстановления фенилгидроксиламином с образованием азоксибензола. Эта реакция по существу подобна присоединению азотистых оснований к карбонильной группе альдегидов и кетонов.

В лабораторных условиях азоксибензол с хорошим выходом получается при восстановлении нитросоединений боргидридом натрия в ДМСО, метилатом натрия в метиловом спирте или старым способом при использовании в качестве восстановителя As 2 O 3 или глюкозы.

Азоксибензол при действии цинка в спиртовом растворе щелочи восстанавливается сначала до азобензола, а при действии избытка цинка далее до гидразобензола.

В синтетической практике производные азоксибензола могут быть восстановлены до азобензола под действием триалкилфосфита в качестве восстановителя. С другой стороны, азобензол легко окисляется до азоксибензола перкислотами.

Азобензол существует в виде цис- и транс- изомеров. При восстановлении азоксибензола получается более стабильный транс-изомер, который при облучении УФ-светом превращается в цис-изомер.

Несимметричные производные азобензола получаются при конденсации нитрозосоединений и первичных ароматических аминов.
При восстановлении ароматических нитросоединений алюмогидридом лития в эфире также образуются азосоединения с выходом, близким к количественному.
Азобензол восстанавливается цинковой пылью и спиртовой щелочью до гидразобензола. Гидразобензол является, таким образом, конечным продуктом восстановления нитробензола металлом в щелочной среде. На воздухе бесцветный гидразобензол легко окисляется до окрашенного в оранжево-красный цвет азобензола. Вместе с тем гидразобензол, также как и азобензол и азоксибензол, восстанавливается до анилина под действием дитионита натрия в воде или хлорида олова (II) в соляной кислоте.
Суммарный процесс восстановления ароматических нитросоединений металлами в кислой и щелочной среде может быть представлен в виде следующей последовательности превращений.
В кислой среде:
В щелочной среде:

В промышленности анилин получают каталитическим восстановлением нитробензола на медном или никелевом катализаторе, который вытеснил старинный способ восстановления нитробензола чугунными стружками в водном растворе хлорного железа и соляной кислоты.
Восстановление нитрогруппы до аминогруппы сульфидом и гидросульфидом натрия в настоящее время имеет значение только для частичного восстановления одной из двух нитрогрупп, например, в м-динитробензоле или 2,4-динитроанилине.

При ступенчатом восстановлении полинитросоединений с помощью сульфида натрия этот неорганический реагент превращается в тетрасульфид натрия, что сопровождается образованием щелочи.
Высокая щелочность среды приводит к образованию азокси- и азосоединений в качестве побочных продуктов. Для того чтобы избежать этого в качестве восстановителя следует использовать гиросульфид натрия, где щелочь не образуется.
| " |
Нитросоедингениями называют производные углеводородов, в которых один или несколько атомов водорода замещены на нитрогруппу -NO 2 . В зависимости от углеводородного радикала, к которому присоединена нитрогруппа, нитросоединения делятся на ароматические и алифатические. Алифатические соединения различают как первичные 1о, вторичные 2 о и третичные 3о, в зависимости от того к 1 о, 2 о или 3о атому углерода присоединена нитрогруппа.
Нитрогруппу -NO2 не следует путать с нитритной группой -ONO. Нитрогруппа имеет следующее строение:
Наличие полного положительного заряда на атоме азота обусловливает наличие у нее сильного -I-эффекта. Наряду с сильным -I-эффектом нитрогруппа обладает сильным -М-эффектом.
Упр. 1. Рассмотрите строение нитрогруппы и ее влияние на направление, и скорость реакции электрофильного замещения в ароматическом ядре.
Способы получения нитросоединений
Практически все способы получения нитросоединений были уже рассмотрены в предыдущих главах. Ароматические нитросоединения получают, как правило, прямым нитрованием аренов и ароматических гетероциклических соединений. Нитроциклогексан в промышленных условиях получают нитрованием циклогексана:

Таким же путем получают и нитрометан, однако в лабораторных условиях его получают из хлоруксусной кислоты в результате реакций (2-5). Ключевой стадией из них является реакция (3), проходящая по механизму SN2.
Хлоруксусная кислота Хлорацетат натрия
Нитроуксусная кислота
Нитрометан
Реакции нитросоединений
Таутомерия алифатических нитросоединений
Вследствие сильных электроноакцепторных свойств нитрогруппы, -атомы водорода обладают повышенной подвижностью и поэтому первичные и вторичные нитросоединения являются С-Н-кислотами. Так, нитрометан является довольно сильной кислотой (pKa 10,2) и в щелочной среде легко превращается в резонансностабилизированный анион:
Нитрометан pKa 10,2 Резонансностабилизированный анион
Упр.2. Напишите реакции (а) нитрометана и (б) нитроциклогексана с водным раствором NaOH.
Конденсация алифатических нитросоединений с альдегидами и кетонами
Нитрогруппа может быть введена в алифатические соединения альдольной реакцией между анионом нитроалкана и альдегидом или кетоном. В нитроалканах -атомы водорода даже более подвижны, чем в альдегидах и кетонах и поэтому они могут вступать с альдегидами и кетонами в реакции присоединения и конденсации предоставляя свои -атомы водорода. С алифатическими альдегидами обычно проходят реакции присоединения, а с ароматическими - исключительно конденсации.
Так, нитрометан присоедняется к циклогексанону,

1-Нитрометилциклогексанол
но конденсируется с бензальдегидом,
В реакции присоединения с формальдегидом участвуют все три атома водорода нитрометана и образуется 2-гидроксиметил-2-нитро-1,3-динитропропан или триметилолнитрометан.
Конденсацией нитрометана с гексаметилентетрамином мы получили 7-нитро-1,3,5-триазаадамантан:

Упр. 3. Напишите реакции формальдегида (а) с нитрометаном и (б) с нитроциклогексаном в щелочной среде.
Восстановление нитросоединений
Нитрогруппу восстанавливают в аминогруппу различными восстановителями (11.3.3). Гидрированием нитробензола под давлением в присутствии никеля Ренея в промышленных условиях получают анилин

В лабораторных условиях вместо водорода можно использовать гидразин, разлагающийся в присутствии никеля Ренея с выделением водорода.

7-нитро-1,3,5-триазаадамантан 7-амино-1,3,5-триазаадамантан
Нитросоединения восстанавливают металлами в кислой среде с последующим подщелачиванием

В зависимости от рН среды и используемого восстановителя могут быть получены различные продукты. В нейтральной и щелочной среде активность обычных восстанавливающих агентов по отношению к нитросоединениям меньше, чем в кислой среде. Характерным примером может служить восстановление нитробензола цинком. В избытке соляной кислоты цинк восстанавливает нитробензол в анилин, в то время как в буферном растворе аммонийхлорида - в фенилгидроксиламин:

В кислой среде арилгидроксиламины подвергаются перегруппировке:

п-Аминофенол используется в качестве проявителя в фотографии. Фенилгидроксиламин далее может быть окислен до нитрозобензола:

Нитрозобензол
Восстановлением нитробензола хлоридом олова (II) получают азобензол, а цинком в щелочной среде - гидразобензол.


Обработкой нитробензола раствором щелочи в метаноле получают азоксибензол, при этом метанол окисляется в муравьиную кислоту.

Известны методы неполного восстановления и нитроалканов. На этом основан один из промышленных методов получения капрона. Нитрованием циклогексана получают нитроциклогексан, который восстановлением переводят в оксим циклогексанона и далее с помощью перегруппировки Бекмана - в капролактам и полиамид - исходное вещество для приготовления волокна - капрона:

Восстановление нитрогруппы продуктов альдольного присоединения (7) является удобным способом получения -аминоспиртов.

1-Нитрометилциклогексанол 1-Аминометилциклогексанол
Использование в качестве восстановителя сероводорода позволяет восстанавливать одну из нитрогрупп в динитроаренах:

м-Динитробензол м-Нитроанилин

2,4-Динитроанилин 4-Нитро-1,2-диаминобензол
Упр.4. Напишите реакции восстановления (а) м-динитробензола оловом в соляной кислоте, (б) м-динитробензола сероводородом, (в) п-нитротолуола цинком в буферном растворе хлорида аммония.
Упр.5. Завершите реакции: