Alcane permanganate de potassium en milieu acide. Oxydation de diverses classes de composés organiques
Institut technologique d'État de Saint-Pétersbourg
(Université technique)
département chimie organique Faculté 4
Groupe 476
Travail de cours
Oxydation des alcènes
Étudiant……………………………………… Rytina A.I.
Conférencier………………………………... Piterskaya Yu.L.
Saint-Pétersbourg
Introduction
1. Époxydation (réaction de N.A. Prilezhaev, 1909)
2. Hydroxylation
2.1anti-Hydroxylation
2.2syn-Hydroxylation
3. Clivage oxydatif des alcènes
4.Ozonolyse
5. Oxydation des alcènes en présence de sels de palladium
Conclusion
Liste des sources utilisées
Introduction
L'oxydation est l'une des transformations les plus importantes et les plus répandues des composés organiques.
En chimie organique, l'oxydation est comprise comme des processus qui conduisent à l'appauvrissement d'un composé en hydrogène ou à son enrichissement en oxygène. Dans ce cas, les électrons sont retirés de la molécule. En conséquence, la réduction est comprise comme le détachement d'une molécule d'oxygène organique ou l'addition d'hydrogène à celle-ci.
Dans les réactions redox, les agents oxydants sont des composés à forte affinité électronique (électrophiles) et les agents réducteurs sont des composés qui ont tendance à donner des électrons (nucléophiles). La facilité d'oxydation d'un composé augmente avec la croissance de sa nucléophilie.
Lors de l'oxydation des composés organiques, en règle générale, il ne se produit pas de transfert complet d'électrons et, par conséquent, de modification de la valence des atomes de carbone. Par conséquent, le concept de degré d'oxydation - la charge conditionnelle d'un atome dans une molécule, calculée sur la base de l'hypothèse que la molécule n'est constituée que d'ions - n'est que conditionnel, formel.
Lors de la compilation des équations des réactions redox, il est nécessaire de déterminer l'agent réducteur, l'agent oxydant et le nombre d'électrons donnés et reçus. En règle générale, les coefficients sont sélectionnés à l'aide de la méthode de l'équilibre électron-ion (méthode de la demi-réaction).
Cette méthode considère la transition des électrons d'un atome ou d'un ion à un autre, en tenant compte de la nature du milieu (acide, alcalin ou neutre) dans lequel la réaction a lieu. Pour égaliser le nombre d'atomes d'oxygène et d'hydrogène, on introduit soit des molécules d'eau et des protons (si le milieu est acide), soit des molécules d'eau et des ions hydroxyde (si le milieu est alcalin).
Ainsi, lors de l'écriture des demi-réactions de réduction et d'oxydation, il faut partir de la composition des ions réellement présents dans la solution. Les substances peu dissociées, peu solubles ou dégagées sous forme de gaz doivent être écrites sous forme moléculaire.
A titre d'exemple, considérons le processus d'oxydation de l'éthylène avec une solution aqueuse diluée de permanganate de potassium (réaction de Wagner). Au cours de cette réaction, l'éthylène est oxydé en éthylène glycol et le permanganate de potassium est réduit en dioxyde de manganèse. Deux hydroxyles 1 sont ajoutés à l'endroit de la double liaison :
3C 2 H 4 + 2KMnO 4 + 4H 2 O → 3C 2 H 6 O 2 + 2MnO 2 + 2KOH
Demi-réaction de réduction : MnO 4 ¯ + 2H 2 O + 3 e → MnO 2 + 4OH ¯ 2
Demi-réaction d'oxydation : C 2 H 4 + 2OH − − 2 e → C 2 H 6 O 2 3
Enfin, nous avons sous forme ionique :
2MnO 4 ¯ + 4H 2 O + 3C 2 H 4 + 6OH ¯ → 2MnO 2 + 8OH ¯ + 3C 2 H 6 O 2
Après avoir effectué les réductions nécessaires de termes semblables, on écrit l'équation sous forme moléculaire :
3C 2 H 4 + 2KMnO 4 + 4 H 2 O \u003d 3C 2 H 6 O 2 + 2MnO 2 + 2KOH.
Caractéristiques de certains agents oxydants
Oxygène
L'oxygène de l'air est largement utilisé dans les procédés technologiques, car c'est l'agent oxydant le moins cher. Mais l'oxydation avec l'oxygène de l'air se heurte à des difficultés liées au contrôle du processus, qui se déroule dans des directions différentes. L'oxydation est généralement effectuée à haute température en présence de catalyseurs.
Ozone
L'ozone O 3 est utilisé pour obtenir des aldéhydes et des cétones, s'il est difficile de les obtenir par d'autres moyens. Le plus souvent, l'ozone est utilisé pour établir la structure des composés insaturés. L'ozone est produit par l'action d'une décharge électrique silencieuse sur l'oxygène. L'un des avantages non négligeables de l'ozonation, par rapport à la chloration, est l'absence de toxines après traitement 2 .
Le permanganate de potassium
Le permanganate de potassium est l'agent oxydant le plus couramment utilisé. Le réactif est soluble dans l'eau (6,0 % à 20 °C), ainsi que dans le méthanol, l'acétone et l'acide acétique. Pour l'oxydation, des solutions aqueuses (parfois d'acétone) de KMnO 4 sont utilisées en milieu neutre, acide ou alcalin. Lors de la réalisation du procédé dans un environnement neutre, des sels de magnésium, d'aluminium sont ajoutés à la masse réactionnelle ou du dioxyde de carbone est traversé pour neutraliser l'hydroxyde de potassium libéré lors de la réaction. La réaction d'oxydation du KMnO 4 en milieu acide est le plus souvent réalisée en présence d'acide sulfurique. L'environnement alcalin lors de l'oxydation est créé par le KOH formé lors de la réaction, ou il est initialement ajouté à la masse réactionnelle. En milieu légèrement alcalin et neutre, KMnO 4 s'oxyde selon l'équation :
KMnO4+ 3 e+ 2H 2 O \u003d K + + MnO 2 + 4OH ¯
en milieu acide :
KMnO4+ 5 e+ 8H + = K + + Mn 2+ + 4H 2 O 3
Le permanganate de potassium est utilisé pour obtenir des 1,2-diols à partir d'alcènes, dans l'oxydation d'alcools primaires, d'aldéhydes et d'alkylarènes en acides carboxyliques, ainsi que pour le clivage oxydatif du squelette carboné au niveau de liaisons multiples.
En pratique, un excès assez important (plus de 100%) de KMnO 4 est généralement utilisé. Cela est dû au fait que, dans des conditions normales, KMnO 4 se décompose partiellement en dioxyde de manganèse avec libération d'O 2 . Se décompose de manière explosive avec H 2 SO 4 concentré lorsqu'il est chauffé en présence d'agents réducteurs; les mélanges de permanganate de potassium avec des substances organiques sont également explosifs 4 .
Peracides
Les acides peracétique et performique sont obtenus en faisant réagir du peroxyde d'hydrogène à 25-90% avec l'acide carboxylique correspondant selon la réaction suivante :
RCOOH + H 2 O 2 \u003d RCOOOH + H 2 O
Dans le cas de l'acide acétique, cet équilibre s'établit relativement lentement et l'acide sulfurique est généralement ajouté comme catalyseur pour accélérer la formation de peracide. L'acide formique est assez fort à lui seul pour fournir un équilibre rapide.
L'acide pertrifluoroacétique, obtenu en mélange avec l'acide trifluoroacétique par réaction de l'anhydride trifluoroacétique avec du peroxyde d'hydrogène à 90%, est un agent oxydant encore plus puissant. De même, l'acide peracétique peut être obtenu à partir d'anhydride acétique et de peroxyde d'hydrogène.
Solide m-l'acide chloroperbenzoïque, car il est relativement sûr à manipuler, assez stable et peut être stocké pendant une longue période.
L'oxydation se produit en raison de l'atome d'oxygène libéré :
RCOOH = RCOOH + [O]
Les peracides sont utilisés pour obtenir des époxydes à partir d'alcènes, ainsi que des lactones à partir de cétones alicycliques.
Peroxyde d'hydrogène
Le peroxyde d'hydrogène est un liquide incolore, miscible avec l'eau, l'éthanol et l'éther diéthylique. Une solution à 30% de H 2 O 2 est appelée perhydrol. Une préparation très concentrée peut réagir de manière explosive avec des substances organiques. Lors du stockage, il se décompose en oxygène et en eau. La persistance du peroxyde d'hydrogène augmente avec la dilution. Pour l'oxydation, des solutions aqueuses de diverses concentrations (de 3 à 90 %) sont utilisées en milieu neutre, acide ou alcalin.
H 2 O 2 \u003d H 2 O + [O]
Par l'action de ce réactif sur des composés carbonylés α,β-insaturés en milieu alcalin, on obtient les époxyaldéhydes et cétones correspondants, on synthétise des peracides par oxydation d'acides carboxyliques en milieu acide. Une solution à 30% de H 2 O 2 dans l'acide acétique oxyde les alcènes en 1,2-diols. Le peroxyde d'hydrogène est utilisé : pour obtenir des peroxydes organiques et inorganiques, perborate et percarbonate de Na ; comme agent oxydant dans les carburants pour fusées ; à réception d'époxydes, d'hydroquinone, de pyrocatéchol, d'éthylène glycol, de glycérine, d'accélérateurs de vulcanisation du groupe thiurame, etc. ; pour blanchir les huiles, les graisses, la fourrure, le cuir, les matières textiles, le papier ; pour nettoyer les matériaux semi-conducteurs au germanium et au silicium ; comme désinfectant pour la neutralisation des eaux usées domestiques et industrielles ; en médecine; comme source d'O 2 dans les sous-marins; H 2 O 2 fait partie du réactif de Fenton (Fe 2 + + H 2 O 2 ), utilisé comme source de radicaux libres OH en synthèse organique 5 .
Tétroxydes de ruthénium et d'osmium
Le tétroxyde d'osmium OsO 4 est une poudre blanche à jaune pâle avec un pf. 40.6ºС ; t.kip. 131.2ºС. Sublime déjà à température ambiante, soluble dans l'eau (7,47 g dans 100 ml à 25ºС), СCl 4 (250 g dans 100 g de solvant à 20ºС). En présence de composés organiques, il devient noir par réduction en OsO 2 6 .
RuO 4 est un prisme jaune doré avec si pl. 25,4ºС, se sublime sensiblement à température ambiante. Peu soluble dans l'eau (2,03 g dans 100 ml à 20ºС), très soluble dans CCl 4 . Un agent oxydant plus fort que OsO 4 . Au-dessus de 100ºС explose. Comme le tétroxyde d'osmium, il a une toxicité élevée et un coût élevé.
Ces agents oxydants sont utilisés pour l'oxydation des alcènes en α-glycols dans des conditions douces.
Dioxiranes
Les plus couramment utilisés sont le diméthyldioxirane et le méthyl(trifluorométhyl)dioxirane.

Les dioxiranes sont le plus souvent obtenus dans situéà partir des cétones correspondantes et KHSO 5 (ou K 2 SO 4 KHSO 4 2KHSO 5 (oxone)) en milieu légèrement alcalin 7 :
Les dioxiranes se distinguent par une réactivité élevée associée à une bonne sélectivité et sont utilisés pour l'oxydation des liaisons C – H non activées dans les alcanes, dans la préparation d'époxydes à partir d'alcènes, pour l'oxydation d'amines, d'oximes, de sulfures, de sulfoxydes, etc.
L'oxydation des alcènes peut avoir lieu dans plusieurs directions :
1) avec la préservation du squelette carboné de la molécule ; c'est ainsi que procèdent l'époxydation et l'hydroxylation de la double liaison, conduisant à la formation de trans- ou cis-glycols vicinaux.
2) avec rupture de la double liaison ; c'est ainsi que se déroulent l'ozonolyse et l'oxydation exhaustive des alcènes, conduisant à la formation de divers types de composés carbonylés et d'acides carboxyliques.
Selon le type d'oxydation, divers agents oxydants sont utilisés.
1. Epoxydation(réaction de N.A. Prilezhaev, 1909)
Les alcènes acycliques et cycliques, lorsqu'ils interagissent avec des peracides (peracides) RCOOOH dans un milieu non polaire et indifférent, forment des époxydes (oxiranes), c'est pourquoi la réaction elle-même est appelée réaction d'époxydation.

Selon la nomenclature moderne UICPA- un cycle à trois chaînons avec un atome d'oxygène est appelé oxirane.
L'époxydation des alcènes doit être considérée comme un processus synchrone et coordonné, qui n'implique pas d'intermédiaires ioniques tels que le cation hydroxyle OH+. En d'autres termes, l'époxydation des alcènes est un processus syn- ajout d'un atome d'oxygène à la double liaison avec préservation complète de la configuration des substituants au niveau de la double liaison. 8

Pour l'époxydation, on a proposé un mécanisme caractéristique des processus concertés.

L'attaque de la double liaison par l'atome d'oxygène du peracide étant également probable de part et d'autre du plan de la double liaison, les oxiranes résultants sont soit méso-formes, ou mélanges d'énantiomères. Les peracides suivants sont utilisés comme agents époxydants : perbenzoïque, m-chlorperbenzoïque, monoperphtalique, peracétique, trifluoroperacétique et performique. Les peracides aromatiques sont utilisés comme réactifs individuels, tandis que les peracides aliphatiques - CH 3 CO 3 H, CF 3 CO 3 H et HCO 3 H ne sont pas isolés dans formulaire individuel, et sont utilisés après leur formation dans l'interaction du peroxyde d'hydrogène à 30% ou 90% et de l'acide carboxylique correspondant. Perbenzoïque et m-l'acide chloroperbenzoïque est obtenu par oxydation de l'acide benzoïque et m-l'acide chlorobenzoïque avec 70 % de peroxyde d'hydrogène dans une solution d'acide méthanesulfonique ou parmi les chlorures d'acide de ces acides et le peroxyde d'hydrogène.


L'acide monoperphtalique est obtenu par un procédé similaire à partir d'anhydride phtalique et de peroxyde d'hydrogène à 30 %.

A l'heure actuelle, l'acide monoperoxyphtalique sous la forme de son sel de Mg trouve de nombreuses applications, ce réactif pouvant être utilisé dans des mélanges de solvants organiques avec de l'eau 9 .
Initialement, les acides perbenzoïques ou monoperphtaliques ont été utilisés pour obtenir des oxiranes (époxydes) 10 :

Actuellement, l'époxydation est le plus souvent utilisée m-acide chloroperbenzoïque. Contrairement aux autres peracides, il est stable pendant le stockage pendant une longue période (jusqu'à 1 an) et sa manipulation est absolument sans danger. Rendements en oxiranes obtenus par oxydation d'alcènes acycliques et cycliques m-chloroperbenzoïque dans une solution de chlorure de méthylène, de chloroforme ou de dioxane sont généralement assez élevés 11 .



Les peracides sont souvent générés directement à partir d'un mélange réactionnel de 90% de peroxyde d'hydrogène et d'acide carboxylique dans du chlorure de méthylène.

Les alcènes à double liaison conjuguée à un groupe carbonyle ou à un autre substituant accepteur sont inactifs et il est préférable d'utiliser des agents oxydants plus puissants pour leur oxydation, comme l'acide trifluoroperacétique obtenu à partir d'anhydride trifluoroacétique et de peroxyde d'hydrogène à 90 % dans du chlorure de méthylène. Les acides peroxycarboxymidiques RC(NH)OOH sont des intermédiaires instables formés dans les réactions des nitriles avec des solutions alcalines de peroxyde d'hydrogène 12 :
L'oxirane le plus simple, l'oxyde d'éthylène, est produit industriellement par oxydation de l'éthylène avec de l'oxygène en présence d'argent comme catalyseur 13 .

2. Hydroxylation
Un certain nombre de réactifs oxydants sont connus, à l'aide desquels, dans des conditions douces, il est possible d'ajouter deux groupes hydroxyle à des alcènes.
La réaction d'hydroxylation des alcènes, se déroulant sous l'action d'une solution froide de permanganate de potassium et accompagnée de sa décoloration, est connue sous le nom de réaction de Wagner (1888). Il a actuellement peu d'utilisation synthétique, car il s'accompagne de la formation d'un nombre important de sous-produits. Cependant, cette réaction peut être utilisée dans l'étude de la structure d'un composé organique comme test qualitatif d'une double liaison. L'hydroxylation du cyclohexène par action d'une solution aqueuse de permanganate de potassium à froid a d'abord été réalisée par V.V. Markovnikov. Actuellement, l'oxyde d'osmium(VIII) est le plus souvent utilisé pour l'hydroxylation des alcènes (réaction de Kriege, 1936) 14 .
Le tétroxyde d'osmium OsO 4 est une substance cristalline incolore, fondant à 40 o C et facilement soluble dans l'éther. Lors de l'oxydation du cyclohexène avec une solution éthérée de ce réactif, il se forme un précipité noir, qui est un ester cyclique de l'acide osmique I 15 :
L'éther est formé à la suite de l'ouverture simultanée de la double liaison carbone-carbone de l'alcène et des deux doubles liaisons de l'oxyde métallique. Cet ester est ensuite hydrolysé en utilisant du sulfite de sodium comme catalyseur. Le produit d'hydrolyse est le cis-cyclohexanediol-1,2(II), dans lequel les hydroxyles sont situés dans la région β, c'est-à-dire devant, et des hydrogènes dans la région α, c'est-à-dire derrière.
2.1 anti-Hydroxylation
Le cycle à trois chaînons des oxiranes s'ouvre facilement sous l'action d'une grande variété de réactifs nucléophiles. L'hydrolyse des oxiranes est catalysée à la fois par des acides et des bases. Dans les deux cas, des diols vicinaux, c'est-à-dire des glycols, se forment. Au cours de la catalyse acide, dans la première étape, la protonation de l'atome d'oxygène de l'oxirane se produit avec la formation d'un cation oxonium cyclique, qui s'ouvre à la suite de l'attaque nucléophile d'une molécule d'eau 16 :

L'étape clé de l'ouverture du cycle, qui détermine la vitesse de l'ensemble du processus, est l'attaque nucléophile de l'eau sur la forme protonée de l'oxirane. Du point de vue du mécanisme, ce processus est similaire à l'ouverture de l'ion bromonium lors de l'attaque nucléophile de l'ion bromure ou d'un autre agent nucléophile. À partir de ces positions, le résultat stéréochimique devrait être la formation transe-glycols dans le clivage des époxydes cycliques. En effet, lors de l'hydrolyse catalysée par un acide de l'oxyde de cyclohexène ou de l'oxyde de cyclopentène, exclusivement transe-1,2-diols.
Ainsi, le processus en deux étapes d'époxydation d'alcène suivie d'une hydrolyse acide de l'époxyde correspond au total à la réaction anti-hydroxylation des alcènes.
Les deux étapes anti-l'hydroxylation des alcènes peut être combinée si l'alcène est traité avec du peroxyde d'hydrogène aqueux à 30-70% dans de l'acide formique ou trifluoroacétique. Ces deux acides sont suffisamment forts pour ouvrir le cycle oxirane.

L'ouverture du cycle oxirane, catalysée par la base, conduit également à la formation de transe-glycols.
Par conséquent, le processus en deux étapes d'époxydation des alcènes suivi d'une hydrolyse alcaline des époxydes est également une réaction anti-hydroxylation des alcènes.
2.2 syn-Hydroxylation
Certains sels et oxydes de métaux de transition dans des états d'oxydation plus élevés sont des réactifs efficaces. syn-hydroxylation de la double liaison d'un alcène, lorsque les deux groupes hydroxyle sont attachés du même côté de la double liaison. L'oxydation des alcènes avec du permanganate de potassium est l'une des méthodes les plus anciennes syn-hydroxylation à double liaison - continue d'être largement utilisée malgré ses limites inhérentes. cis-1,2-cyclohexanediol a d'abord été obtenu par V.V. Markovnikov en 1878 par hydroxylation du cyclohexène avec une solution aqueuse de permanganate de potassium à 0 0 C.

Cette méthode a été développée plus avant dans les travaux du scientifique russe E.E. Wagner donc syn-l'hydroxylation des alcènes sous l'action d'une solution aqueuse de permanganate de potassium est appelée réaction de Wagner. Le permanganate de potassium est un agent oxydant puissant qui peut non seulement hydroxyler la double liaison, mais aussi cliver le diol vicinal résultant. Afin d'éviter autant que possible une dégradation supplémentaire des glycols, les conditions de réaction doivent être soigneusement contrôlées. Les rendements en glycol sont généralement faibles (30-60%). Meilleurs résultats sont obtenus par hydroxylation d'alcènes en milieu légèrement alcalin (рН~8 9) à 0-5 0 С avec une solution aqueuse diluée à 1% de KMnO 4 17 .


Initialement, lorsque les alcènes sont oxydés avec du permanganate de potassium, un ester d'acide permanganique cyclique se forme, qui est immédiatement hydrolysé en un diol vicinal.

L'ester cyclique de l'acide permanganique n'a pas été isolé comme intermédiaire, mais sa formation découle d'expériences avec du permanganate de potassium marqué 18 O : les deux atomes d'oxygène du glycol se révèlent marqués lors de l'oxydation de l'alcène KMn 18 O 4 . Cela signifie que les deux atomes d'oxygène sont transférés de l'agent oxydant et non du solvant - l'eau, ce qui est en bon accord avec le mécanisme proposé.
Une autre méthode syn-l'hydroxylation des alcènes sous l'action de l'oxyde d'osmium (VIII) OsO 4 a été proposée par R. Krige en 1936. Le tétroxyde d'osmium est une substance cristalline incolore, volatile, facilement soluble dans l'éther, le dioxane, la pyridine et d'autres solvants organiques. Lorsque le tétroxyde d'osmium réagit avec des alcènes dans l'éther ou le dioxane, un précipité noir de l'ester cyclique d'acide osmique se forme - l'osmate, qui peut être facilement isolé individuellement. L'addition de OsO 4 à la double liaison est nettement accélérée en solution de pyridine. La décomposition des osmates en glycols vicinaux est réalisée par l'action d'une solution aqueuse d'hydrosulfite de sodium ou d'hydrogène sulfuré.

Sorties du produit syn-l'hydroxylation des alcènes dans cette méthode est beaucoup plus élevée que lors de l'utilisation du permanganate comme agent oxydant. Un avantage important de la méthode de Krige est l'absence de produits du clivage oxydatif des alcènes, caractéristique de l'oxydation du permanganate 18 .
 19
19

Le tétroxyde d'osmium est un réactif très coûteux et difficile à obtenir, en plus d'être toxique. Par conséquent, l'oxyde d'osmium (VIII) est utilisé dans la synthèse de petites quantités de substances difficiles à atteindre afin d'obtenir le rendement en diol le plus élevé. Afin de simplifier syn-hydroxylation des alcènes sous l'action de OsO 4 une technique a été développée qui permet de n'utiliser que des quantités catalytiques de ce réactif. L'hydroxylation des alcènes est réalisée à l'aide de peroxyde d'hydrogène en présence d'OsO 4, par exemple :

En conclusion, nous pouvons donner la relation stéréochimique entre l'alcène cis- ou transe-configuration et configuration du diol vicinal résultant, qui peut être cis- ou transe-isomère, érythro- ou tréo-former, méso- ou D,L-forme en fonction des substituants dans l'alcène 20 :

Des relations stéréochimiques similaires sont observées dans d'autres réactions syn- ou anti- additions de liaisons multiples d'hydrogène, d'halogénures d'hydrogène, d'eau, d'halogènes, d'hydrures de bore et d'autres réactifs.
3. Clivage oxydatif alcènes
Lors de l'oxydation d'alcènes avec une solution aqueuse alcaline de permanganate de potassium lorsqu'elle est chauffée ou avec une solution de KMnO 4 dans de l'acide sulfurique aqueux, ainsi que lors de l'oxydation d'alcènes avec une solution d'oxyde de chrome (VI) CrO 3 dans de l'acide acétique ou dichromate de potassium et acide sulfurique, le glycol initialement formé subit une dégradation oxydative. Le résultat final est la division du squelette carboné au site de la double liaison et la formation de cétones et/ou d'acides carboxyliques comme produits finaux, en fonction des substituants sur la double liaison. Si les deux atomes de carbone au niveau de la double liaison ne contiennent qu'un seul groupe alkyle, le produit final de l'oxydation exhaustive sera un mélange d'acides carboxyliques, l'alcène tétrasubstitué au niveau de la double liaison est oxydé en deux cétones. Les alcènes monosubstitués avec une double liaison terminale sont clivés en acide carboxylique et en dioxyde de carbone 21 .

 22
22
En raison des faibles rendements en acides carboxyliques et en cétones, les réactions d'oxydation exhaustive des alcènes dans la version classique n'ont pas trouvé une large application et étaient auparavant principalement utilisées pour déterminer la structure de l'alcène initial à partir des produits de l'oxydation destructrice. Actuellement, l'oxydation des alcènes (R-CH=CH-R et R-CH=CH2) en acides carboxyliques (RCOOH) avec du permanganate ou du dichromate de potassium est réalisée dans des conditions de catalyse par transfert de phase. Les rendements en acides carboxyliques dépassent dans ce cas 90 %.

4. Ozonolyse des alcènes
La réaction des alcènes avec l'ozone est la méthode la plus importante pour le clivage oxydatif des alcènes au niveau de la double liaison. Pendant de nombreuses décennies, cette réaction a servi de méthode principale pour déterminer la structure de l'hydrocarbure initial et a également trouvé une application dans la synthèse de divers composés carbonylés. La réaction de l'alcène avec l'ozone est réalisée en faisant passer un courant d'environ 5% de mélange d'ozone et d'oxygène dans une solution d'alcène dans du chlorure de méthylène ou de l'acétate d'éthyle à -80 0 -100 0 C. La fin de la réaction est contrôlée par un test d'ozone libre avec de l'iodure de potassium. Le mécanisme de cette réaction particulière et complexe a été établi principalement grâce aux travaux de Krige. Le premier produit de la cycloaddition 1,3-dipolaire à la double liaison est ce que l'on appelle le molozonide (1,2,3-trioxolane). Ce produit est instable et se décompose ensuite spontanément avec ouverture du cycle et formation d'ozonide normal (1,2,4-trioxolane) 23 comme produit final.
 24
24
Il est maintenant généralement admis que la transformation du molozonide en ozonide ordinaire se produit par le mécanisme de scission-recombinaison. Le mollozonide subit une ouverture spontanée du cycle 1,2,3-trioxolane instable avec la formation d'un composé carbonyle et d'un ion bipolaire, qui réagissent ensuite l'un avec l'autre également selon le schéma de cycloaddition 1,3-dipolaire.

Le schéma donné du réarrangement du molozonide en ozonide normal est confirmé par le fait que si un autre composé carbonyle est présent en tant qu '"intercepteur" de l'ion bipolaire dans le mélange réactionnel avant la formation complète de l'ozonide, alors le soi-disant " ozonide mixte" se forme. Par exemple, dans l'ozonisation cis-stilbène en présence de benzaldéhyde marqué par l'isotope 18 O, le marqueur fait partie de l'éther, et non le pont peroxyde de l'ozonide :
Ce résultat est en bon accord avec la formation d'un ozonide mixte lors de la recombinaison d'un ion bipolaire avec du benzaldéhyde marqué :

Les ozonides sont des composés hautement instables qui se décomposent de manière explosive. Ils ne sont pas isolés individuellement, mais éclatés sous l'action d'une grande variété de régents. Il faut faire la distinction entre clivage réducteur et clivage oxydatif. Au cours de l'hydrolyse, les ozonides sont lentement divisés en composés carbonylés et en peroxyde d'hydrogène. Le peroxyde d'hydrogène oxyde les aldéhydes en acides carboxyliques. C'est la décomposition dite oxydative des ozonides :

Ainsi, lors de la décomposition oxydative des ozonides, acides carboxyliques et (ou) des cétones selon la structure de l'alcène de départ. L'oxygène de l'air, le peroxyde d'hydrogène, les peracides ou l'hydroxyde d'argent peuvent être utilisés comme agents oxydants. Le plus souvent dans la pratique de la synthèse, on utilise à cette fin le peroxyde d'hydrogène dans l'acide acétique ou formique, ainsi que le peroxyde d'hydrogène en milieu alcalin.


En pratique, la méthode de décomposition oxydative des ozonides est principalement utilisée pour obtenir des acides carboxyliques.
Plus important est le clivage réducteur des ozonides. Les agents réducteurs les plus couramment utilisés sont le zinc et l'acide acétique, la triphénylphosphine ou le sulfure de diméthyle. Dans ce cas, les produits finaux de l'ozonolyse sont des aldéhydes ou des cétones, selon la structure de l'alcène de départ.


 25
25
D'après les exemples ci-dessus, on peut voir qu'un alcène tétra-substitué au niveau d'une double liaison forme deux cétones pendant l'ozonolyse et la décomposition réductrice ultérieure de l'ozonide, tandis qu'un alcène tri-substitué donne une cétone et un aldéhyde. Un alcène symétrique disubstitué forme deux aldéhydes au cours de l'ozonolyse, et les alcènes avec une liaison terminale forment un aldéhyde et un formaldéhyde.
Une modification intéressante de l'ozonolyse est la méthode où le borohydrure de sodium est utilisé comme agent réducteur d'ozonide.Dans ce cas, les produits de réaction finaux sont des alcools primaires ou secondaires formés lors de la réduction des aldéhydes et des xtones, respectivement 26 .


L'ozonolyse des alcènes est un processus complexe, long et explosif qui nécessite l'utilisation d'équipements spéciaux. Pour cette raison, d'autres méthodes ont été développées pour le clivage oxydatif des alcènes en composés carbonylés et en acides carboxyliques, qui remplacent avec succès la réaction d'ozonolyse dans la pratique synthétique.
L'une des méthodes modernes de préparation pour la destruction oxydative des alcènes a été proposée en 1955 par R. Lemieux. Cette méthode est basée sur l'hydroxylation des alcènes avec du permanganate de potassium, suivie du clivage du glycol vicinal avec du periodate de sodium NaIO 4 à pH ~ 7 8. Le periodate lui-même n'interagit pas avec l'alcène. Les produits de ce clivage oxydatif en deux étapes sont des cétones ou des acides carboxyliques, puisque les aldéhydes sont également oxydés en acides carboxyliques dans ces conditions. Dans la méthode Lemieux, le problème laborieux de la séparation de l'un des produits de réaction, le dioxyde de manganèse, ne se pose pas, puisque le dioxyde et le manganate sont à nouveau oxydés avec du periodate en un ion permanganate. Cela permet d'utiliser uniquement des quantités catalytiques de permanganate de potassium. Voici quelques exemples typiques du clivage oxydatif d'alcènes par la méthode de Lemieux.

Le citronellol, un alcool qui fait partie des huiles de rose, de géranium et de citron, est oxydé avec un mélange de permanganate de potassium et de periodate de sodium dans de l'acétone aqueuse à 5 10 0 C en acide 6-hydroxy-4-méthylhexanecarboxylique avec un rendement quantitatif.

Une autre variante de cette méthode utilise des quantités catalytiques de tétroxyde d'osmium au lieu de permanganate de potassium (Lemieux & Johnson 1956). Un avantage particulier de l'association OsO 4 et NaIO 4 est qu'elle permet de stopper l'oxydation au stade aldéhydique. Le tétroxyde d'osmium s'ajoute à la double liaison de l'alcène pour former l'osmate, qui est oxydé par le periodate de sodium en composés carbonylés avec la régénération du tétroxyde d'osmium.

A la place du tétroxyde d'osmium, on peut également utiliser le tétroxyde de ruthénium RuO 4 . La dégradation oxydative Lemieux-Johnson des alcènes conduit aux mêmes produits que l'ozonolyse avec clivage réducteur des ozonides.

En termes caractéristiques de la chimie organique moderne, cela signifie que la combinaison OsO 4 -NaIO 4 est équivalent synthétique ozonolyse des alcènes suivie d'un clivage réducteur. De même, l'oxydation des alcènes avec un mélange de permanganate et de periodate est l'équivalent synthétique de l'ozonolyse avec dégradation oxydative des ozonides.
Ainsi, l'oxydation des alcènes n'est pas seulement un ensemble de méthodes de préparation pour l'obtention d'alcools, d'époxydes, de diols, d'aldéhydes, de cétones et d'acides carboxyliques, c'est aussi l'une des voies possibles pour établir la structure de l'alcène de départ. Ainsi, selon le résultat de la dégradation oxydative de l'alcène, on peut déterminer la position de la double liaison dans la molécule, tandis que le résultat stéréochimique syn- ou anti- l'hydroxylation d'un alcène permet de tirer une conclusion sur sa géométrie.
5. Oxydation des alcènes en présence de sels de palladium
L'oxydation des alcènes en composés carbonylés à l'aide de chlorure de palladium a fait l'objet de nombreuses recherches, principalement en raison de l'importance pour l'industrie de la réaction de production d'acétaldéhyde à partir d'éthylène (procédé Wacker). Au cours de l'oxydation, le chlorure de palladium est réduit en palladium, et un tel procédé aurait un intérêt limité si ce n'était du fait que le chlorure de palladium coûteux peut être utilisé en quantités catalytiques en présence d'un second oxydant, le plus souvent du chlorure de cuivre (II) , qui oxyde le palladium en palladium.(II), tout en étant lui-même réduit en cuivre(I).La réoxydation du cuivre(I) en cuivre(II) peut être effectuée avec de l'oxygène atmosphérique, de sorte que le procédé global est très attractif en tant que méthode d'oxydation industrielle 27 .
L'éthylène est facilement oxydé en acétaldéhyde 28 :
Mécanisme suggéré 29 :
La réaction se déroule en milieu acide, ne s'accompagne pas d'une modification du nombre d'atomes de carbone dans une molécule d'éthylène, et est actuellement la principale source d'acétaldéhyde dans l'industrie.
L'oxydation des homologues de l'éthylène dans les mêmes conditions se produit au niveau de l'atome de carbone le moins hydrogéné de la double liaison avec formation de cétones. En particulier, lorsque le propène est oxydé, on obtient de l'acétone, et lorsque le cyclohexène est oxydé, on obtient de la cyclohexanone.
Conclusion
La réaction d'oxydation est un groupe important de réactions de double liaison. De manière générale, les réactions d'oxydation occupent une place particulière en chimie organique. Lors de l'examen de ces réactions, il faut tenir compte non seulement de la nature du composé organique à oxyder, mais aussi de la nature de l'agent oxydant, de la présence ou non d'un catalyseur, du milieu dans lequel se déroule la réaction, etc.
Il est donc souvent nécessaire de mémoriser les agents oxydants et leurs conditions d'utilisation pour obtenir le produit d'oxydation recherché. Par exemple, la réaction d'oxydation des alcènes avec une solution diluée de permanganate de potassium conduit à la formation de diols (glycols), tandis qu'une solution concentrée de permanganate de potassium détruit la molécule d'alcène au niveau de la double liaison avec formation de produits contenant de l'oxygène. Ainsi, le même agent oxydant dans des environnements différents donne des produits d'oxydation différents.
Parmi les réactions d'oxydation, la réaction d'ozonation considérée par nous est considérée comme la plus importante, ce qui permet d'établir la structure de l'alcène initial à partir des produits finaux. Dans cet article, les principales réactions d'oxydation des alcènes et des catalyseurs utilisés au cours des processus oxydatifs ont été examinées.
Liste des sources utilisées :
1) Reutov O. A. Chimie organique. En 4 parties. Ch. 1.-M. : BINOM. Laboratoire des connaissances, 2004.-567p.
2) Remy G. Cours de chimie inorganique. T. 1. M. : Maison d'édition de littérature étrangère, 1963. - 920 p.
3) D.V. Kazakov, A.I. Voloshin, V.P. Kazakov, V.V. Shereschovets, N.N. Kabalnova. Chimie et chimiluminescence des dioxiranes. Moscou : Nauka, 1999
4) Traven VF . Chimie Organique : Manuel pour les Lycées : En 2 volumes / VF Traven. - M. : CPI "Akademkniga", 2004. - T. 1
5) Haynes A. Méthodes d'oxydation des composés organiques. Alcanes, alcènes, alcynes et arènes. M. : Mir, 1988.
6) Morrison R., Boyd R. "Chimie organique" M. : Mir, 1974
7) Shabarov Yu.S. "Chimie organique" partie 1 M. : Chimie 1994
8) Petrov A.A., Balyan H.V., Troshchenko A.T. Chimie organique : un manuel pour les universités - 5e éd., révisée. Et supplémentaire - Saint-Pétersbourg: "Ivan Fedorov", 2002.-624 p.
9) L Plume, M. Fizer Organic Chemistry.Advanced Course.Volume 1-M., "Chimie", 1969, 688 p.
10) Plotnikov V.F., Saint-Pétersbourg Yu.L. Alcènes Tutoriel.-SPb: SPbGTI(TU)-2003.-20 p.
11) Neiland O. Ya. Chapitre II. Alcènes // Chimie Organique : Proc. pour chim. les universités. - M.: "L'école supérieure", 1990.
12) Mars D. Chimie organique : En 4 volumes / Per. de l'anglais. - M. : Mir, 1987. - T. 4. 470 p.
13) Terney A. Chimie organique moderne : en 2 volumes - M. : Mir, 1981.
14) Hauptman Z., Grefe Yu., Remane H. "Chimie organique" M. : Khimiya, 1979
15) Dryuk V.G., Malinovsky M.S. "Cours de chimie organique" Kiev: école Vishcha 1987
16) http://www.chemistry.ssu.samara.ru/chem2/u442.htm
17) http://www.xumuk.ru/
1 Petrov A.A., Balyan H.V., Troshchenko A.T. Chimie organique : Manuel pour les universités, p.85
26 26 Traven Résumé >> Chimie
CH2 + 3O2 2CO2 + 2H2O b) À oxydation alcènes une solution diluée de permanganate de potassium se forme... sur une double liaison. c) Quand dur oxydation alcènes solution bouillante de permanganate de potassium dans de l'acide ...
Théorie de la chimie générale avec des éléments de méthodologie d'enseignement
Aide-mémoire >> ChimieLa méthode d'obtention des composés carbonylés - oxydation les alcools. En tant qu'agent oxydant, vous pouvez ... rendement ~ 90%) 5. Oxydation alcènes. Les aldéhydes et les cétones sont oxydation les hydrocarbures de la série de l'éthylène... ne sont pas hydratés. 7. Réactions oxydation. Aldéhydes et cétones...
Oxiranes (époxydes)
Résumé >> Chimie...) 3,4-époxy-1-butène Procédés de préparation d'oxiranes Oxydation alcènes peracides La méthode la plus utilisée... acide 1,2-époxycyclo-m-chlorobenzoïque acide hexanique Oxydation alcènes peracides organiques s'accompagne de l'ajout d'oxygène...
Informations générales sur les alcools. Polyols
Résumé >> Chimie...) peut être obtenu oxydation alcènes permanganate de potassium ou tétrokisdom .... Décrire le mécanisme de la réaction Oxydation polyols Oxydation l'éthylène glycol procède différemment, ... mécanismes : (m 21) Périodique oxydation la glycérine conduit à la formation de formaldéhyde...
Ce matériel peut être difficile à maîtriser avec l'auto-apprentissage, en raison de la grande quantité d'informations, de nombreuses nuances, de toutes sortes de MAIS et SI. Lire attentivement!
De quoi sera-t-il question exactement ?
En plus de l'oxydation complète (combustion), certaines classes de composés organiques sont caractérisées par des réactions d'oxydation partielle, alors qu'elles sont converties en d'autres classes.
Il existe des agents oxydants spécifiques pour chaque classe : CuO (pour les alcools), Cu (OH) 2 et OH (pour les aldéhydes) et autres.
Mais il existe deux agents oxydants classiques, qui, pour ainsi dire, sont universels pour de nombreuses classes.
C'est du permanganate de potassium - KMnO 4. Et le dichromate de potassium (dichromate) - K 2 Cr 2 O 7. Ces substances sont des agents oxydants puissants dus respectivement au manganèse à l'état d'oxydation +7 et au chrome à l'état d'oxydation +6.
Les réactions avec ces agents oxydants sont assez courantes, mais il n'existe nulle part de guide holistique sur la manière de choisir les produits de telles réactions.
En pratique, de nombreux facteurs influent sur le déroulement de la réaction (température, milieu, concentration des réactifs, etc.). Souvent, un mélange de produits est obtenu. Par conséquent, il est presque impossible de prédire le produit qui se forme.
Mais ce n'est pas bon pour l'examen d'État unifié : là, vous ne pouvez pas écrire "peut-être ceci, ou ceci, ou autre chose, ou un mélange de produits". Il faut des précisions.
Les compilateurs des missions ont investi une certaine logique, un certain principe selon lequel un certain produit doit être écrit. Malheureusement, ils n'ont partagé avec personne.
Cette question dans la plupart des manuels est plutôt glissante contournée : deux ou trois réactions sont données à titre d'exemple.
Je présente dans cet article ce que l'on peut appeler les résultats d'une étude-analyse des tâches USE. La logique et les principes de compilation des réactions d'oxydation avec le permanganate et le dichromate ont été décryptés avec une assez grande précision (conformément aux normes USE). A propos de tout dans l'ordre.
Détermination du degré d'oxydation.
Premièrement, lorsqu'il s'agit de réactions redox, il y a toujours un agent oxydant et un agent réducteur.
L'agent oxydant est le manganèse dans le permanganate ou le chrome dans le dichromate, l'agent réducteur est des atomes dans l'organique (à savoir, des atomes de carbone).
Il ne suffit pas de définir les produits, il faut égaliser la réaction. Pour l'égalisation, la méthode de la balance électronique est traditionnellement utilisée. Pour appliquer cette méthode, il est nécessaire de déterminer les états d'oxydation des agents réducteurs et des agents oxydants avant et après la réaction.
À substances inorganiques on connaît le degré d'oxydation dès la 9e année :
Mais en bio, probablement, en 9e année, ils n'étaient pas déterminés. Par conséquent, avant d'apprendre à écrire OVR en chimie organique, vous devez apprendre à déterminer le degré d'oxydation du carbone dans les substances organiques. Cela se fait un peu différemment qu'en chimie inorganique.
Le carbone a un état d'oxydation maximum de +4, un minimum de -4. Et il peut montrer n'importe quel degré d'oxydation de cet intervalle : -4, -3, -2, -1, 0, +1, +2, +3, +4.
Vous devez d'abord vous rappeler ce qu'est un état d'oxydation.
L'état d'oxydation est la charge conditionnelle qui se produit sur un atome, en supposant que les paires d'électrons sont complètement décalées vers l'atome le plus électronégatif.
Par conséquent, l'état d'oxydation est déterminé par le nombre de paires d'électrons déplacées: s'il est déplacé vers un atome donné, il acquiert une charge négative en excès (-), s'il provient d'un atome, il acquiert une charge en excès (+) . En principe, c'est toute la théorie qu'il faut connaître pour déterminer l'état d'oxydation d'un atome de carbone.
Pour déterminer le degré d'oxydation d'un atome de carbone particulier dans un composé, nous devons considérer CHACUNE de ses liaisons et voir dans quelle direction la paire d'électrons se déplacera et quelle charge excessive (+ ou -) en résultera sur l'atome de carbone .
Regardons des exemples spécifiques :
Au carbone trois liaisons hydrogène. Carbone et hydrogène - lequel est le plus électronégatif ? Carbone, puis, le long de ces trois liaisons, la paire d'électrons se déplacera vers le carbone. Le carbone prend une charge négative de chaque hydrogène : il s'avère -3
La quatrième liaison est avec le chlore. Carbone et chlore - lequel est le plus électronégatif ? Chlore, ce qui signifie que sur cette liaison, la paire d'électrons se déplacera vers le chlore. Le carbone a une charge positive +1.
Ensuite, il vous suffit d'ajouter : -3 + 1 = -2. L'état d'oxydation de cet atome de carbone est -2.

Déterminons l'état d'oxydation de chaque atome de carbone :

Le carbone a trois liaisons avec l'hydrogène. Carbone et hydrogène - lequel est le plus électronégatif ? Carbone, puis, le long de ces trois liaisons, la paire d'électrons se déplacera vers le carbone. Le carbone prend une charge négative de chaque hydrogène : il s'avère -3
Et une liaison de plus avec un autre carbone. Carbone et autre carbone - leur électronégativité est égale, il n'y a donc pas de déplacement de la paire d'électrons (la liaison n'est pas polaire). 

Cet atome a deux liaisons avec un atome d'oxygène et une autre liaison avec un autre atome d'oxygène (faisant partie du groupe OH). Plus d'atomes d'oxygène électronégatifs dans trois liaisons tirent une paire d'électrons du carbone, et le carbone a une charge +3.
Par la quatrième liaison, le carbone est relié à un autre carbone, comme nous l'avons déjà dit, la paire d'électrons ne se déplace pas le long de cette liaison.


Le carbone est lié aux atomes d'hydrogène par deux liaisons. Le carbone, comme plus électronégatif, tire une paire d'électrons pour chaque liaison avec l'hydrogène, acquiert une charge de -2.
Une double liaison carbone est liée à un atome d'oxygène. L'oxygène le plus électronégatif attire une paire d'électrons pour chaque liaison. Ensemble, deux paires d'électrons sont extraites du carbone. Le carbone acquiert une charge de +2.
Ensemble, il s'avère +2 -2 = 0.

Déterminons l'état d'oxydation de cet atome de carbone :
Une triple liaison avec un azote plus électronégatif donne au carbone une charge de +3 ; il n'y a pas de déplacement de la paire d'électrons en raison de la liaison avec le carbone.

Oxydation au permanganate.
Qu'adviendra-t-il du permanganate ?
La réaction redox avec le permanganate peut se dérouler dans différents environnements (neutre, alcalin, acide). Et cela dépend du milieu, de la manière exacte dont la réaction se déroulera et des produits formés dans ce cas.
Elle peut donc aller dans trois directions :

Le permanganate, étant un agent oxydant, est réduit. Voici les produits de sa récupération :
- milieu acide.
Le milieu est acidifié avec de l'acide sulfurique (H 2 SO 4 ). Le manganèse est réduit à l'état d'oxydation +2. Et les produits de récupération seront :
KMnO 4 + H 2 SO 4 → MnSO 4 + K 2 SO 4 + H 2 O
- Environnement alcalin.
Pour créer un environnement alcalin, un alcali assez concentré (KOH) est ajouté. Le manganèse est réduit à un état d'oxydation de +6. Produits de récupération
KMnO 4 + KOH → K 2 MnO 4 + H 2 O
- Environnement neutre(et légèrement alcalin).
En milieu neutre, en plus du permanganate, l'eau entre également dans la réaction (que nous écrivons du côté gauche de l'équation), le manganèse sera réduit à +4 (MnO 2), les produits de réduction seront :
KMnO 4 + H 2 O → MnO 2 + KOH
Et en milieu légèrement alcalin (en présence d'une solution de KOH peu concentrée) :
KMnO 4 + KOH → MnO 2 + H 2 O

Qu'adviendra-t-il des matières organiques?
La première chose à apprendre, c'est que tout commence par l'alcool ! C'est la première étape de l'oxydation. Le carbone auquel le groupe hydroxyle est attaché subit une oxydation.
Lorsqu'il est oxydé, l'atome de carbone "acquiert" une liaison avec l'oxygène. Par conséquent, lorsqu'ils écrivent le schéma de la réaction d'oxydation, ils écrivent [O] au-dessus de la flèche :
alcool primaire oxydé d'abord en aldéhyde, puis en acide carboxylique :

Oxydation alcool secondaire pauses dans la deuxième étape. Puisque le carbone est au milieu, une cétone se forme, pas un aldéhyde (l'atome de carbone du groupe cétone ne peut plus physiquement former de liaison avec le groupe hydroxyle) :

Cétones, alcools tertiaires Et acides carboxyliques n'est plus oxydé

Le processus d'oxydation se déroule par étapes - tant qu'il y a où oxyder et que toutes les conditions sont réunies pour cela - la réaction se poursuit. Tout aboutit à un produit qui ne s'oxyde pas dans des conditions données : un alcool tertiaire, une cétone ou un acide.
Il convient de noter les étapes d'oxydation du méthanol. Il est d'abord oxydé en aldéhyde correspondant, puis en acide correspondant :

Une caractéristique de ce produit (acide formique) est que le carbone du groupe carboxyle est lié à l'hydrogène, et si vous regardez attentivement, vous pouvez voir qu'il ne s'agit que d'un groupe aldéhyde :

Et le groupe aldéhyde, comme nous l'avons découvert plus tôt, est oxydé davantage en carboxyle :

Avez-vous reconnu la substance résultante ? Sa formule brute est H 2 CO 3 . Ce acide carbonique, qui se décompose en dioxyde de carbone et en eau :
H2CO3 → H2O + CO2
Par conséquent, le méthanol, l'aldéhyde formique et acide formique(dues au groupe aldéhyde) sont oxydées en dioxyde de carbone.
légère oxydation.
Une légère oxydation est oxydation sans fort chauffage en milieu neutre ou faiblement alcalin (0 s'écrit au dessus de la réaction ° ou 20 °) .
Il est important de se rappeler que les alcools ne s'oxydent pas dans des conditions douces. Par conséquent, s'ils se forment, l'oxydation s'arrête sur eux. Quelles substances entreront dans une réaction d'oxydation douce?
- Contenant une double liaison C=C (réaction de Wagner).
Dans ce cas, la liaison π se rompt et « repose » sur les liaisons libérées le long du groupe hydroxyle. Il s'avère que l'alcool dihydrique:

Écrivons la réaction d'oxydation douce de l'éthylène (éthène). Écrivons les substances initiales et prédisons les produits. En même temps, nous n'écrivons pas encore H 2 O et KOH : ils peuvent apparaître à la fois à droite et à gauche de l'équation. Et nous déterminons immédiatement les états d'oxydation des substances impliquées dans l'OVR :

Faisons une balance électronique (on veut dire qu'il y a deux ou deux atomes de carbone de l'agent réducteur, ils sont oxydés séparément) :

Fixons les coefficients :

A la fin ajouter les produits manquants (H 2 O et KOH). Il n'y a pas assez de potassium à droite - cela signifie que l'alcali sera à droite. Nous mettons un coefficient devant. Il n'y a pas assez d'hydrogène à gauche, donc l'eau est à gauche. On met un coefficient devant :

Faisons de même avec le propylène (propène):


Le cycloalcène est souvent glissé. Ne le laissez pas vous confondre. C'est un hydrocarbure régulier avec une double liaison :

Partout où se trouve cette double liaison, l'oxydation se déroulera de la même manière :

- contenant un groupe aldéhyde.
Le groupe aldéhyde est plus réactif (réagit plus facilement) que le groupe alcool. Par conséquent, l'aldéhyde s'oxydera. Avant acide :

Prenons l'exemple de l'acétaldéhyde (éthanal). Écrivons les réactifs et les produits et organisons les états d'oxydation. Faisons un bilan et mettons les coefficients devant l'agent réducteur et l'agent oxydant :

En milieu neutre et légèrement alcalin, le déroulement de la réaction sera légèrement différent.
Dans un environnement neutre, comme on s'en souvient, on écrit eau sur le côté gauche de l'équation, et alcali sur le côté droit de l'équation (formé lors de la réaction) :

Dans ce cas, dans le même mélange, l'acide et l'alcali sont proches. La neutralisation a lieu.

Ils ne peuvent exister côte à côte et réagir, le sel se forme :
De plus, si nous regardons les coefficients de l'équation, nous comprendrons que les acides sont 3 moles et les alcalis sont 2 moles. 2 moles d'alcali ne peuvent neutraliser que 2 moles d'acide (il se forme 2 moles de sel). Et il reste une mole d'acide. L'équation finale sera donc :

Dans un environnement légèrement alcalin, l'alcali est en excès - il est ajouté avant la réaction, donc tout l'acide est neutralisé :

Une situation similaire se présente dans l'oxydation du méthanal. Il, comme on s'en souvient, est oxydé en dioxyde de carbone:

Il faut garder à l'esprit que le monoxyde de carbone (IV) CO 2 est acide. Et réagira avec l'alcali. Et puisque l'acide carbonique est dibasique, un sel acide et un sel moyen peuvent être formés. Cela dépend du rapport entre l'alcali et le dioxyde de carbone :
Si l'alcali est lié au dioxyde de carbone comme 2: 1, alors il y aura un sel moyen :
Ou l'alcali peut être beaucoup plus (plus de deux fois). Si c'est plus de deux fois, alors le reste de l'alcali restera :
3KOH + CO2 → K2CO3 + H2O + KOH
Cela se produira dans un environnement alcalin (où il y a un excès d'alcali, car il a été ajouté au mélange réactionnel avant la réaction) ou dans un environnement neutre, lorsqu'il se forme beaucoup d'alcali.
Mais si l'alcali est lié au dioxyde de carbone comme 1: 1, alors il y aura un sel acide :
KOH + CO2 → KHCO3
S'il y a plus de dioxyde de carbone que nécessaire, alors il reste en excès :
KOH + 2CO 2 → KHCO 3 + CO 2
Ce sera dans un environnement neutre si peu d'alcali se forme.
Nous écrivons les substances de départ, les produits, établissons un bilan, notons les états d'oxydation devant l'oxydant, l'agent réducteur et les produits qui en sont formés:

Dans un environnement neutre, un alcali (4KOH) se formera à droite :

Nous devons maintenant comprendre ce qui se formera lorsque trois moles de CO 2 et quatre moles d'alcali interagissent.
3CO2 + 4KOH → 3KHCO3 + KOH
KHCO 3 + KOH → K 2 CO 3 + H 2 O
Alors ça se passe comme ça :
3CO2 + 4KOH → 2KHCO3 + K2CO3 + H2O
Par conséquent, sur le côté droit de l'équation, nous écrivons deux moles d'hydrocarbure et une mole de carbonate:

Et dans un environnement légèrement alcalin, il n'y a pas de tels problèmes: du fait qu'il y a un excès d'alcali, un sel moyen se formera:

La même chose se produira avec l'oxydation de l'aldéhyde de l'acide oxalique :

Comme dans l'exemple précédent, un acide dibasique se forme, et selon l'équation, 4 moles d'alcali doivent être obtenues (puisque 4 moles de permanganate).
Dans un environnement neutre, encore une fois, tout l'alcali n'est pas suffisant pour neutraliser complètement tout l'acide.
Trois moles d'alcali vont former un sel acide, il reste une mole d'alcali :
3HOOC–COOH + 4KOH → 3KOOC–COOH + KOH
Et cette mole d'alcali entre en interaction avec une mole de sel acide :
KOOC–COOH + KOH → KOOC–COOK + H2O
Ça se passe comme ça :
3HOOC–COOH + 4KOH → 2KOOC–COOH + KOOC–COOK + H2O
Équation finale :

En milieu faiblement alcalin, un sel moyen se forme du fait d'un excès d'alcali :

- contenant une triple liaisonC≡ C.
Vous souvenez-vous de ce qui s'est passé lors de l'oxydation douce des composés à double liaison ? Si vous ne vous en souvenez pas, faites défiler vers l'arrière - rappelez-vous.
La liaison π se rompt, se fixe aux atomes de carbone au niveau du groupe hydroxyle. Ici même principe. N'oubliez pas qu'il y a deux liaisons pi dans une triple liaison. Tout d'abord, cela se produit à la première liaison π :
Puis sur une autre liaison π :

Une structure dans laquelle un atome de carbone a deux groupes hydroxyle est extrêmement instable. Quand quelque chose est instable en chimie, il a tendance à "tomber" quelque chose. L'eau tombe, comme ceci :

Il en résulte un groupe carbonyle.
Prenons des exemples :
Éthine (acétylène). Considérez les étapes d'oxydation de cette substance:

Fractionnement de l'eau :




Comme dans l'exemple précédent, dans un mélange réactionnel, acide et alcalin. La neutralisation se produit - du sel se forme. Comme le montre le coefficient devant le permanganate alcalin, il y aura 8 moles, c'est-à-dire qu'il suffit amplement de neutraliser l'acide. Équation finale :

Considérez l'oxydation du butyne-2 :

Fractionnement de l'eau :

Aucun acide ne se forme ici, il n'est donc pas nécessaire de s'amuser avec la neutralisation.
Équation de réaction :

Ces différences (entre l'oxydation du carbone en bord et en milieu de chaîne) sont clairement mises en évidence par l'exemple du pentyne :

Fractionnement de l'eau :

Il s'avère une substance d'une structure intéressante:

Le groupe aldéhyde continue de s'oxyder :
Inscrivons les matières premières, les produits, déterminons le degré d'oxydation, établissons un bilan, inscrivons les coefficients devant l'oxydant et l'agent réducteur:

L'alcali devrait former 2 mol (puisque le coefficient devant le permanganate est de 2), par conséquent, tout l'acide est neutralisé :

Oxydation dure.
L'oxydation dure est l'oxydation aigre, fortement alcalin environnement. Et aussi, en neutre (ou légèrement alcalin), mais lorsqu'il est chauffé.
En milieu acide, elles sont aussi parfois chauffées. Mais pour que l'oxydation dure ne se déroule pas dans un environnement acide, le chauffage est une condition préalable.
Quelles substances subiront une oxydation sévère? (D'abord, nous analyserons uniquement dans un environnement acide - puis nous ajouterons les nuances qui apparaissent lors de l'oxydation dans un environnement fortement alcalin et neutre ou légèrement alcalin (lorsqu'il est chauffé)).
Avec une oxydation dure, le processus va au maximum. Tant qu'il y a quelque chose à oxyder, l'oxydation continue.
- Alcools. Aldéhydes.
Considérons l'oxydation de l'éthanol. Progressivement, il s'oxyde en acide :

Nous écrivons l'équation. Nous notons les substances de départ, les produits OVR, notons les états d'oxydation, établissons un bilan. Égaliser la réaction :

Si la réaction est effectuée au point d'ébullition de l'aldéhyde, lorsqu'il se forme, il s'évapore (s'envole) du mélange réactionnel sans avoir le temps de s'oxyder davantage. Le même effet peut être obtenu dans des conditions très douces (faible chaleur). Dans ce cas, nous écrivons aldéhyde en tant que produit :

Considérons l'oxydation de l'alcool secondaire en utilisant l'exemple du propanol-2. Comme déjà mentionné, l'oxydation se termine à la deuxième étape (la formation d'un composé carbonylé). Puisqu'une cétone est formée, qui n'est pas oxydée. Équation de réaction :

Considérez l'oxydation des aldéhydes en termes d'éthanal. Il s'oxyde également en acide :

Équation de réaction :

Le méthanal et le méthanol, comme mentionné précédemment, sont oxydés en dioxyde de carbone :

Métalal :

- Contenant plusieurs liaisons.
Dans ce cas, la chaîne se rompt le long de la liaison multiple. Et les atomes qui l'ont formé subissent une oxydation (acquièrent une liaison avec l'oxygène). Oxyder au maximum.
Lorsqu'une double liaison est rompue, des composés carbonylés sont formés à partir de fragments (dans le schéma ci-dessous: d'un fragment - aldéhyde, de l'autre - cétone)

![]()
Analysons l'oxydation du pentène-2 :

Oxydation des « scraps » :

Il s'avère que deux acides se forment. Notez les matières premières et les produits. Déterminons les états d'oxydation des atomes qui le modifient, établissons un bilan, égalisons la réaction : 
Lors de la compilation de la balance électronique, nous voulons dire qu'il y a deux ou deux atomes de carbone de l'agent réducteur, ils sont oxydés séparément :

L'acide ne se formera pas toujours. Considérons, par exemple, l'oxydation du 2-méthylbutène :

Équation de réaction :

Absolument le même principe dans l'oxydation des composés à triple liaison (seule l'oxydation se produit immédiatement avec la formation d'un acide, sans formation intermédiaire d'un aldéhyde):

Équation de réaction :

Lorsqu'une liaison multiple est située exactement au milieu, on n'obtient pas deux produits, mais un. Étant donné que les "déchets" sont les mêmes et qu'ils sont oxydés en les mêmes produits :

Équation de réaction :

- Acide double corona.
Il existe un acide dans lequel les groupes carboxyle (couronnes) sont reliés les uns aux autres :
Il s'agit de l'acide oxalique. Deux couronnes côte à côte ont du mal à s'entendre. Il est certainement stable dans des conditions normales. Mais du fait qu'il a deux groupes carboxyle reliés l'un à l'autre, il est moins stable que les autres acides carboxyliques.
Et par conséquent, dans des conditions particulièrement difficiles, il peut être oxydé. Il y a une rupture dans la connexion entre les "deux couronnes":

Équation de réaction :

- Homologues du benzène (et leurs dérivés).
Le benzène lui-même ne s'oxyde pas, car l'aromaticité rend cette structure très stable. 
Mais ses homologues sont oxydés. Dans ce cas, le circuit se casse également, l'essentiel est de savoir exactement où. Certains principes s'appliquent :
- Le cycle benzénique lui-même n'est pas détruit et reste intact jusqu'à la fin, la liaison est rompue dans le radical.
- L'atome directement lié au cycle benzénique est oxydé. Si après cela la chaîne carbonée dans le radical continue, alors l'écart sera après lui.


Analysons l'oxydation du méthylbenzène. Là, un atome de carbone du radical est oxydé : 
Équation de réaction :

Analysons l'oxydation de l'isobutylbenzène :

Équation de réaction :

Analysons l'oxydation du sec-butylbenzène :

Équation de réaction :

Lors de l'oxydation d'homologues du benzène (et de dérivés d'homologues) avec plusieurs radicaux, deux à trois acides aromatiques basiques et plus se forment. Par exemple, l'oxydation du 1,2-diméthylbenzène :

Les dérivés d'homologues du benzène (dans lesquels le cycle benzénique a des radicaux non hydrocarbonés) sont oxydés de la même manière. Un autre groupe fonctionnel sur le cycle benzénique n'interfère pas :


Total. Algorithme "comment écrire la réaction d'oxydation dure avec du permanganate dans un environnement acide":
- Notez les matières premières (matières organiques + KMnO 4 + H 2 SO 4).
- Notez les produits d'oxydation organique (les composés contenant de l'alcool, des groupes aldéhyde, des liaisons multiples, ainsi que des homologues du benzène seront oxydés).
- Enregistrer le produit de réduction du permanganate (MnSO 4 + K 2 SO 4 + H 2 O).
- Déterminer le degré d'oxydation chez les participants OVR. Dressez un bilan. Notez les coefficients pour l'agent oxydant et l'agent réducteur, ainsi que pour les substances qui en sont formées.
- Ensuite, il est recommandé de calculer le nombre d'anions sulfate du côté droit de l'équation, conformément à cela, placez le coefficient devant l'acide sulfurique à gauche.
- A la fin, placez le coefficient devant l'eau.
Oxydation sévère en milieu fortement alcalin et en milieu neutre ou faiblement alcalin (lorsqu'il est chauffé).
Ces réactions sont beaucoup moins fréquentes. On peut dire que de telles réactions sont exotiques. Et comme il sied à toutes les réactions exotiques, celles-ci ont été les plus controversées.
L'oxydation dure est également difficile en Afrique, de sorte que les matières organiques sont oxydées de la même manière que dans un environnement acide.
Séparément, nous n'analyserons pas les réactions pour chaque classe, puisque le principe général a déjà été énoncé plus haut. Nous n'analyserons que les nuances.
Environnement fortement alcalin :
En milieu fortement alcalin, le permanganate est réduit à un état d'oxydation de +6 (manganate de potassium) :
KMnO 4 + KOH → K 2 MnO 4 .
Dans un environnement fortement alcalin, il y a toujours un excès d'alcali, par conséquent, une neutralisation complète aura lieu : si du dioxyde de carbone se forme, il y aura un carbonate, si un acide se forme, il y aura un sel (si l'acide est polybasique - un sel moyen).
Par exemple, l'oxydation du propène :

Oxydation de l'éthylbenzène :

Légèrement alcalin ou neutre lorsqu'il est chauffé :
Ici aussi, la possibilité de neutralisation doit toujours être prise en compte.
Si l'oxydation se déroule dans un environnement neutre et qu'un composé acide (acide ou dioxyde de carbone) se forme, l'alcali résultant neutralisera ce composé acide. Mais l'alcali n'est pas toujours suffisant pour neutraliser complètement l'acide.
Lorsque les aldéhydes sont oxydés, par exemple, cela ne suffit pas (l'oxydation se déroulera de la même manière que dans des conditions douces - la température accélérera simplement la réaction). Par conséquent, du sel et de l'acide sont formés (en gros, restant en excès).
Nous en avons discuté lorsque nous avons discuté de l'oxydation douce des aldéhydes.
Par conséquent, si vous avez de l'acide dans un environnement neutre, vous devez voir attentivement s'il suffit de neutraliser tout l'acide. Une attention particulière doit être portée à la neutralisation des acides polybasiques.
Dans un environnement faiblement alcalin, en raison d'une quantité suffisante d'alcali, seuls des sels moyens se forment, car il y a un excès d'alcali.
En règle générale, l'alcali lors de l'oxydation dans un environnement neutre est tout à fait suffisant. Et l'équation de réaction qui en milieu neutre, qu'en milieu légèrement alcalin sera la même.
Par exemple, considérons l'oxydation de l'éthylbenzène :


L'alcali suffit à neutraliser complètement les composés acides résultants, même l'excès restera:

3 moles d'alcali sont consommées - 1 reste.
Équation finale :

Cette réaction en milieu neutre et légèrement alcalin se déroulera de la même manière (en milieu légèrement alcalin il n'y a pas d'alcali à gauche, mais cela ne veut pas dire qu'il n'existe pas, il n'entre tout simplement pas en réaction).
Réactions redox impliquant du dichromate de potassium (bichromate).
Le bichromate n'a pas une telle variété de réactions d'oxydation organique dans l'examen.
L'oxydation au bichromate n'est généralement effectuée qu'en milieu acide. Dans le même temps, le chrome est restauré à +3. Produits de récupération :
L'oxydation sera dure. La réaction sera très similaire à l'oxydation du permanganate. Les mêmes substances seront oxydées qui sont oxydées par le permanganate dans un environnement acide, les mêmes produits seront formés.
Jetons un coup d'œil à certaines des réactions.
Considérez l'oxydation de l'alcool. Si l'oxydation est effectuée au point d'ébullition de l'aldéhyde, alors il sortira de leur mélange réactionnel sans s'oxyder :

Sinon, l'alcool peut être directement oxydé en acide.
L'aldéhyde produit lors de la réaction précédente peut être "capté" et amené à s'oxyder en acide :

Oxydation du cyclohexanol. Le cyclohexanol est un alcool secondaire, il se forme donc une cétone :

S'il est difficile de déterminer les états d'oxydation des atomes de carbone à l'aide de cette formule, vous pouvez écrire sur le brouillon :

Équation de réaction :

Considérons l'oxydation du cyclopentène.
La double liaison se rompt (le cycle s'ouvre), les atomes qui l'ont formée s'oxydent au maximum (dans ce cas, au groupe carboxyle) :


Certaines caractéristiques de l'oxydation dans l'UTILISATION avec lesquelles nous ne sommes pas entièrement d'accord.
Ces "règles", principes et réactions qui seront discutés dans cette section, nous ne les considérons pas tout à fait corrects. Ils contredisent non seulement l'état réel des choses (la chimie en tant que science), mais aussi la logique interne programme scolaire et l'UTILISATION en particulier.
Mais néanmoins, nous sommes obligés de donner ce matériel sous la forme que l'USE exige.
On parle d'oxydation DURE.
Rappelez-vous comment les homologues du benzène et leurs dérivés sont oxydés dans des conditions difficiles ? Tous les radicaux sont terminés - des groupes carboxyle sont formés. Les chutes sont déjà oxydées "indépendamment":

Donc, si soudainement un groupe hydroxyle, ou une liaison multiple, apparaît sur le radical, vous devez oublier qu'il y a un cycle benzénique là-bas. La réaction ira UNIQUEMENT le long de ce groupe fonctionnel (ou liaison multiple).
Le groupe fonctionnel et la liaison multiple sont plus importants que le cycle benzénique.

Analysons l'oxydation de chaque substance:
Première matière :

Il ne faut pas faire attention au fait qu'il y a un cycle benzénique. Du point de vue de l'examen, ce n'est que de l'alcool secondaire. Les alcools secondaires sont oxydés en cétones et les cétones ne sont plus oxydées :

Que cette substance soit oxydée avec du bichromate :

Deuxième substance :

Cette substance est oxydée, tout comme un composé à double liaison (on ne fait pas attention au cycle benzénique) :

Laissez-le s'oxyder dans du permanganate neutre lorsqu'il est chauffé :

L'alcali résultant est suffisant pour neutraliser complètement le dioxyde de carbone :
2KOH + CO2 → K2CO3 + H2O
Équation finale :

Oxydation de la troisième substance :

Laisser l'oxydation se poursuivre avec du permanganate de potassium en milieu acide :

Oxydation de la quatrième substance :

Laissez-le s'oxyder dans un environnement fortement alcalin. L'équation de la réaction sera :

Et enfin, voici comment le vinylbenzène est oxydé :

Et il s'oxyde en acide benzoïque, il faut garder à l'esprit que, selon la logique de l'examen d'État unifié, il s'oxyde de cette façon non pas parce qu'il s'agit d'un dérivé du benzène. Parce qu'il contient une double liaison.
Conclusion.
C'est tout ce que vous devez savoir sur les réactions redox impliquant le permanganate et le dichromate dans les matières organiques.
Ne soyez pas surpris si certains des points évoqués dans cet article, vous entendez pour la première fois. Comme déjà mentionné, ce sujet est très vaste et controversé. Et malgré cela, pour une raison quelconque, très peu d'attention y est accordée.
Comme vous l'avez peut-être vu, deux ou trois réactions n'expliquent pas tous les schémas de ces réactions. Ici, vous avez besoin d'une approche intégrée et d'une explication détaillée de tous les points. Malheureusement, dans les manuels et sur les ressources Internet, le sujet n'est pas entièrement divulgué, ou pas du tout divulgué.
J'ai essayé d'éliminer ces lacunes et lacunes et de considérer ce sujet dans son intégralité, et non en partie. J'espère avoir réussi.
Merci de votre attention, bonne chance à vous ! Succès dans le développement science chimique et passer des examens !
Comme déjà mentionné, l'oxydation d'une matière organique est l'introduction d'oxygène dans sa composition et (ou) l'élimination d'hydrogène. La récupération est le processus inverse (introduction d'hydrogène et élimination d'oxygène). Compte tenu de la composition des alcanes (СnH2n+2), on peut conclure qu'ils sont incapables de participer aux réactions de réduction, mais ils peuvent participer aux réactions d'oxydation.
Les alcanes sont des composés à faible degré d'oxydation du carbone et, selon les conditions de réaction, ils peuvent être oxydés pour former divers composés.
Aux températures ordinaires, les alcanes ne réagissent pas même avec des agents oxydants puissants (H2Cr2O7, KMnO4, etc.). Lorsqu'ils sont introduits dans une flamme nue, les alcanes brûlent. En même temps, dans un excès d'oxygène, ils sont complètement oxydés en CO2, où le carbone a le degré le plus élevé oxydation +4 et eau. La combustion des hydrocarbures entraîne la rupture de tous Connexions CC et C-H et s'accompagne d'un dégagement de chaleur important (réaction exothermique).
Il est généralement admis que le mécanisme d'oxydation des alcanes comprend un processus de chaîne radicalaire, puisque l'oxygène lui-même n'est pas très réactif, pour extraire un atome d'hydrogène d'un alcane, il faut une particule qui initiera la formation d'un radical alkyle qui réagir avec l'oxygène, donnant un radical peroxy. Le radical peroxy peut alors extraire un atome d'hydrogène d'une autre molécule d'alcane pour former un hydroperoxyde d'alkyle et un radical.

Il est possible d'oxyder les alcanes avec l'oxygène atmosphérique à 100-150 ° C en présence d'un catalyseur - l'acétate de manganèse, cette réaction est utilisée dans l'industrie. L'oxydation se produit lorsqu'un courant d'air est soufflé à travers de la paraffine fondue contenant un sel de manganèse.
Parce que à la suite de la réaction, un mélange d'acides se forme, puis ils sont séparés de la paraffine n'ayant pas réagi par dissolution dans un alcali aqueux, puis neutralisés avec un acide minéral.
Directement dans l'industrie, cette méthode est utilisée pour obtenir de l'acide acétique à partir de n-butane :
Oxydation des alcènes
Les réactions d'oxydation des alcènes sont divisées en deux groupes : 1) les réactions dans lesquelles le squelette carboné est préservé, 2) les réactions de destruction oxydative du squelette carboné de la molécule le long de la double liaison.
Réactions d'oxydation des alcènes avec préservation du squelette carboné
1. Époxydation (réaction de Prilezhaev)
Les alcènes acycliques et cycliques, lorsqu'ils interagissent avec des peracides dans un milieu non polaire, forment des époxydes (oxiranes).
De plus, les oxiranes peuvent être obtenus par oxydation d'alcènes avec des hydroperoxydes en présence de catalyseurs contenant du molybdène, du tungstène et du vanadium :

L'oxirane le plus simple, l'oxyde d'éthylène, est produit industriellement par oxydation de l'éthylène avec de l'oxygène en présence d'argent ou d'oxyde d'argent comme catalyseur.

2. anti-hydroxylation (hydrolyse des époxydes)
L'hydrolyse acide (ou alcaline) des époxydes conduit à l'ouverture du cycle des oxydes avec formation de transdiols.

Dans la première étape, la protonation de l'atome d'oxygène de l'époxyde se produit avec la formation d'un cation oxonium cyclique, qui s'ouvre à la suite de l'attaque nucléophile de la molécule d'eau.
L'ouverture du cycle époxy catalysée par une base conduit également à la formation de trans-glycols.

3. syn-hydroxylation
L'une des plus anciennes méthodes d'oxydation des alcènes est la réaction de Wagner (oxydation au permanganate de potassium). Initialement, lors de l'oxydation, un ester de permanganate cyclique se forme, qui est hydrolysé en un diol vicinal :

En plus de la réaction de Wagner, il existe une autre méthode de syn-hydroxylation des alcènes sous l'action de l'oxyde d'osmium (VIII), qui a été proposée par Krige. Sous l'action du tétroxyde d'osmium sur un alcène dans l'éther ou le dioxane, un précipité noir de l'ester cyclique de l'acide osmique se forme - l'osmate. Cependant, l'addition d'OsO4 à la liaison multiple est nettement accélérée dans la pyridine. Le précipité noir d'osmate qui en résulte est facilement décomposé par l'action d'une solution aqueuse d'hydrosulfite de sodium :

Le permanganate de potassium ou l'oxyde d'osmium (VIII) oxydent l'alcène en cis-1,2-diol.
Clivage oxydatif des alcènes
Le clivage oxydatif des alcènes comprend les réactions de leur interaction avec le permanganate de potassium dans l'acide alcalin ou sulfurique, ainsi que l'oxydation avec une solution de trioxyde de chrome dans l'acide acétique ou le dichromate de potassium et l'acide sulfurique. Le résultat final de telles transformations est la division du squelette carboné au site de la double liaison et la formation d'acides carboxyliques ou de cétones.
Les alcènes monosubstitués avec une double liaison terminale sont clivés en un acide carboxylique et du dioxyde de carbone :

Si les deux atomes de carbone de la double liaison ne contiennent qu'un seul groupe alkyle, un mélange d'acides carboxyliques se forme :

Mais si un alcène tétrasubstitué par une double liaison est une cétone :

La réaction d'ozonolyse des alcènes a acquis une importance préparative beaucoup plus grande. Pendant de nombreuses décennies, cette réaction a servi de méthode principale pour déterminer la structure de l'alcène de départ. Cette réaction est réalisée en faisant passer un courant d'une solution d'ozone dans l'oxygène, une solution d'alcène dans le chlorure de méthylène ou l'acétate d'éthyle à -80...-100°C. Le mécanisme de cette réaction a été établi par Krige :


Les ozonides sont des composés instables qui se décomposent lors d'une explosion. Il existe deux voies de décomposition des ozonides - oxydative et réductrice.
Au cours de l'hydrolyse, les ozonides sont divisés en composés carbonylés et en peroxyde d'hydrogène. Le peroxyde d'hydrogène oxyde les aldéhydes en acides carboxyliques - c'est la décomposition oxydative :

La séparation réductrice des ozonides est beaucoup plus importante. Les produits d'ozonolyse sont des aldéhydes ou des cétones, selon la structure de l'alcène de départ :
En plus des méthodes ci-dessus, il existe une autre méthode proposée en 1955 par Lemieux :
Dans la méthode Lemieux, il n'y a pas de procédures fastidieuses pour séparer le dioxyde de manganèse, puisque le dioxyde et le manganate sont à nouveau oxydés avec du periodate en ion permanganate. Cela permet d'utiliser uniquement des quantités catalytiques de permanganate de potassium.
4.5. Oxydation des alcènes
Il convient de diviser les réactions d'oxydation des alcènes en deux grands groupes : les réactions de préservation du squelette carboné et les réactions de destruction oxydative du squelette carboné de la molécule le long de la double liaison. Le premier groupe de réactions comprend l'époxydation, ainsi que l'hydroxylation, conduisant à la formation de diols vicinaux (glycols). Dans le cas des alcènes cycliques, l'hydroxylation forme vicinal transe- ou cis-diols. Un autre groupe comprend l'ozonolyse et les réactions d'oxydation exhaustive des alcènes, conduisant à la formation de divers types de composés carbonylés et d'acides carboxyliques.
4.5.a. Réactions d'oxydation des alcènes avec préservation du squelette carboné
1. Époxydation (réaction de N.A. Prilezhaev, 1909)
Les alcènes acycliques et cycliques, lorsqu'ils interagissent avec des peracides (peracides) RCOOOH dans un milieu non polaire et indifférent, forment des époxydes (oxiranes), c'est pourquoi la réaction elle-même est appelée réaction d'époxydation.
Selon la nomenclature moderne UICPA- un cycle à trois chaînons avec un atome d'oxygène est appelé oxirane.
L'époxydation des alcènes doit être considérée comme un processus synchrone et coordonné, qui n'implique pas d'intermédiaires ioniques tels que le cation hydroxyle OH+. En d'autres termes, l'époxydation des alcènes est un processus syn- ajout d'un atome d'oxygène à la double liaison avec préservation complète de la configuration des substituants au niveau de la double liaison.
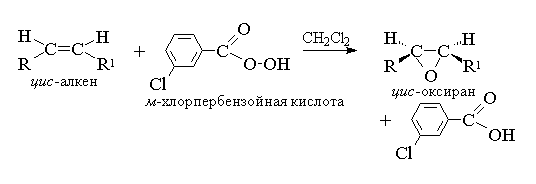

Pour l'époxydation, on a proposé un mécanisme caractéristique des processus concertés.

L'attaque de la double liaison par l'atome d'oxygène du peracide étant également probable de part et d'autre du plan de la double liaison, les oxiranes résultants sont soit méso-formes, ou mélanges d'énantiomères. Les peracides suivants sont utilisés comme agents époxydants : perbenzoïque, m-chlorperbenzoïque, monoperphtalique, peracétique, trifluoroperacétique et performique. Les peracides aromatiques sont utilisés comme réactifs individuels, tandis que les peracides aliphatiques - CH 3 CO 3 H, CF 3 CO 3 H et HCO 3 H ne sont pas isolés individuellement, mais sont utilisés après leur formation dans l'interaction de 30% ou 90% de peroxyde d'hydrogène et l'acide carboxylique correspondant. Perbenzoïque et m-l'acide chloroperbenzoïque est obtenu par oxydation de l'acide benzoïque et m-l'acide chlorobenzoïque avec 70 % de peroxyde d'hydrogène dans une solution d'acide méthanesulfonique ou parmi les chlorures d'acide de ces acides et le peroxyde d'hydrogène.


L'acide monoperphtalique est obtenu par un procédé similaire à partir d'anhydride phtalique et de peroxyde d'hydrogène à 30 %.

Initialement, les acides perbenzoïques ou monoperphtaliques étaient utilisés pour obtenir des oxiranes (époxydes) :

Actuellement, l'époxydation est le plus souvent utilisée m-acide chloroperbenzoïque. Contrairement aux autres peracides, il est stable pendant le stockage pendant une longue période (jusqu'à 1 an) et sa manipulation est absolument sans danger. Rendements en oxiranes obtenus par oxydation d'alcènes acycliques et cycliques m-chloroperbenzoïque dans une solution de chlorure de méthylène, de chloroforme ou de dioxane sont généralement assez élevés.



Les peracides sont souvent générés directement à partir d'un mélange réactionnel de 90% de peroxyde d'hydrogène et d'acide carboxylique dans du chlorure de méthylène.


Les alcènes à double liaison conjuguée à un groupe carbonyle ou à un autre substituant accepteur sont inactifs et il est préférable d'utiliser des agents oxydants plus puissants pour leur oxydation, comme l'acide trifluoroperacétique obtenu à partir d'anhydride trifluoroacétique et de peroxyde d'hydrogène à 90 % dans du chlorure de méthylène. L'oxirane le plus simple, l'oxyde d'éthylène, est produit industriellement par oxydation de l'éthylène avec de l'oxygène en présence d'argent comme catalyseur.

2. anti-Hydroxylation
Le cycle à trois chaînons des oxiranes s'ouvre facilement sous l'action d'une grande variété de réactifs nucléophiles. Ces réactions seront discutées en détail dans la section sur les éthers acycliques et cycliques. Ici, seule l'hydrolyse des oxiranes sera considérée. L'hydrolyse des oxiranes est catalysée à la fois par des acides et des bases. Dans les deux cas, des diols vicinaux, c'est-à-dire des glycols, se forment. Lors de la catalyse acide, dans la première étape, la protonation de l'atome d'oxygène de l'oxirane se produit avec la formation d'un cation oxonium cyclique, qui s'ouvre suite à l'attaque nucléophile d'une molécule d'eau :

L'étape clé de l'ouverture du cycle, qui détermine la vitesse de l'ensemble du processus, est l'attaque nucléophile de l'eau sur la forme protonée de l'oxirane. Du point de vue du mécanisme, ce processus est similaire à l'ouverture de l'ion bromonium lors de l'attaque nucléophile de l'ion bromure ou d'un autre agent nucléophile. À partir de ces positions, le résultat stéréochimique devrait être la formation transe-glycols dans le clivage des époxydes cycliques. En effet, lors de l'hydrolyse catalysée par un acide de l'oxyde de cyclohexène ou de l'oxyde de cyclopentène, exclusivement transe-1,2-diols.

Ainsi, le processus en deux étapes d'époxydation d'alcène suivie d'une hydrolyse acide de l'époxyde correspond au total à la réaction anti-hydroxylation des alcènes.
Les deux étapes anti-l'hydroxylation des alcènes peut être combinée si l'alcène est traité avec du peroxyde d'hydrogène aqueux à 30-70% dans de l'acide formique ou trifluoroacétique. Ces deux acides sont suffisamment forts pour ouvrir le cycle oxirane.

L'ouverture du cycle oxirane, catalysée par la base, conduit également à la formation de transe-glycols.

Par conséquent, le processus en deux étapes d'époxydation des alcènes suivi d'une hydrolyse alcaline des époxydes est également une réaction anti-hydroxylation des alcènes.
3. syn-Hydroxylation
Certains sels et oxydes de métaux de transition dans des états d'oxydation plus élevés sont des réactifs efficaces. syn-hydroxylation de la double liaison d'un alcène, lorsque les deux groupes hydroxyle sont attachés du même côté de la double liaison. L'oxydation des alcènes avec du permanganate de potassium est l'une des méthodes les plus anciennes syn-hydroxylation à double liaison - continue d'être largement utilisée malgré ses limites inhérentes. cis-1,2-cyclohexanediol a d'abord été obtenu par V.V. Markovnikov en 1878 par hydroxylation du cyclohexène avec une solution aqueuse de permanganate de potassium à 0 0 C.

Cette méthode a été développée plus avant dans les travaux du scientifique russe E.E. Wagner donc syn-l'hydroxylation des alcènes sous l'action d'une solution aqueuse de permanganate de potassium est appelée réaction de Wagner. Le permanganate de potassium est un agent oxydant puissant qui peut non seulement hydroxyler la double liaison, mais aussi cliver le diol vicinal résultant. Afin d'éviter autant que possible une dégradation supplémentaire des glycols, les conditions de réaction doivent être soigneusement contrôlées. Les rendements en glycol sont généralement faibles (30-60%). Les meilleurs résultats sont obtenus par hydroxylation des alcènes en milieu légèrement alcalin (рН~8 9) à 0-5 0 С avec une solution aqueuse diluée à 1% de KMnO 4 .


Initialement, lorsque les alcènes sont oxydés avec du permanganate de potassium, un ester d'acide permanganique cyclique se forme, qui est immédiatement hydrolysé en un diol vicinal.

L'ester cyclique de l'acide permanganique n'a pas été isolé comme intermédiaire, mais sa formation découle d'expériences avec du permanganate de potassium marqué 18 O : les deux atomes d'oxygène du glycol se révèlent marqués lors de l'oxydation de l'alcène KMn 18 O 4 . Cela signifie que les deux atomes d'oxygène sont transférés de l'agent oxydant et non du solvant - l'eau, ce qui est en bon accord avec le mécanisme proposé.
Une autre méthode syn-l'hydroxylation des alcènes sous l'action de l'oxyde d'osmium (VIII) OsO 4 a été proposée par R. Krige en 1936. Le tétroxyde d'osmium est une substance cristalline incolore, volatile, facilement soluble dans l'éther, le dioxane, la pyridine et d'autres solvants organiques. Lorsque le tétroxyde d'osmium réagit avec des alcènes dans l'éther ou le dioxane, un précipité noir de l'ester cyclique d'acide osmique se forme - l'osmate, qui peut être facilement isolé individuellement. L'addition de OsO 4 à la double liaison est nettement accélérée en solution de pyridine. La décomposition des osmates en glycols vicinaux est réalisée par l'action d'une solution aqueuse d'hydrosulfite de sodium ou d'hydrogène sulfuré.

Sorties du produit syn-l'hydroxylation des alcènes dans cette méthode est beaucoup plus élevée que lors de l'utilisation du permanganate comme agent oxydant. Un avantage important de la méthode de Krige est l'absence de produits de clivage oxydatif des alcènes, caractéristique de l'oxydation du permanganate.


Le tétroxyde d'osmium est un réactif très coûteux et difficile à obtenir, en plus d'être toxique. Par conséquent, l'oxyde d'osmium (VIII) est utilisé dans la synthèse de petites quantités de substances difficiles à atteindre afin d'obtenir le rendement en diol le plus élevé. Afin de simplifier syn-hydroxylation des alcènes sous l'action de OsO 4 une technique a été développée qui permet de n'utiliser que des quantités catalytiques de ce réactif. L'hydroxylation des alcènes est réalisée à l'aide de peroxyde d'hydrogène en présence d'OsO 4, par exemple :

Pour conclure cette section, nous présentons les relations stéréochimiques entre l'alcène cis- ou transe-configuration et configuration du diol vicinal résultant, qui peut être cis- ou transe-isomère, érythro- ou tréo-former, méso- ou D,L-forme en fonction des substituants dans l'alcène :

Des relations stéréochimiques similaires sont observées dans d'autres réactions syn- ou anti- additions de liaisons multiples d'hydrogène, d'halogénures d'hydrogène, d'eau, d'halogènes, d'hydrures de bore et d'autres réactifs.
Établir des équations de réactions redox impliquant des substances organiques
DANS lien avec l'introduction comme seule forme certification finale anciens élèves lycée unifié Examen d'état(UTILISATION) et transition lycée pour l'enseignement spécialisé, la préparation des lycéens à accomplir les tâches les plus « chères » de la partie « C » en termes de points devient de plus en plus importante. UTILISER l'essai en chimie. Malgré le fait que les cinq tâches de la partie "C" sont considérées comme différentes : les propriétés chimiques des substances inorganiques, les chaînes de transformations des composés organiques, les tâches de calcul, toutes sont dans une certaine mesure liées aux réactions redox (ORR). Si les connaissances de base de la théorie OVR sont maîtrisées, il est alors possible de terminer correctement les première et deuxième tâches dans leur intégralité, et la troisième - partiellement. À notre avis, une part importante du succès de la mise en œuvre de la partie "C" réside précisément dans cela. L'expérience montre que si, en étudiant la chimie inorganique, les étudiants maîtrisent assez bien les tâches d'écriture d'équations OVR, des tâches similaires en chimie organique leur causent de grandes difficultés. Par conséquent, tout au long de l'étude de l'ensemble du cours de chimie organique dans des classes spécialisées, nous essayons de développer chez les lycéens les compétences de compilation des équations OVR.
En étudiant caractéristiques comparatives des composés inorganiques et organiques, nous initions les étudiants à l'utilisation de l'état d'oxydation (s.o.) (en chimie organique, principalement le carbone) et comment le déterminer :
1) calcul de la moyenne s.d. le carbone dans une molécule de matière organique ;
2) définition de s.d. chaque atome de carbone.
Nous clarifions dans quels cas il est préférable d'utiliser l'une ou l'autre méthode.
L'article a été publié avec le soutien de la société "GEO-Engineering", qui présente des produits sur le marché sous la marque "ProfKresla". Le domaine d'activité de l'entreprise est la production, la vente et l'installation de fauteuils et de chaises pour différentes salles. Le haut professionnalisme des employés et nos propres installations de production nous permettent de mettre en œuvre rapidement et efficacement des projets de toute complexité. Tous les produits de la marque "ProfKresla", qu'il s'agisse de chaises de théâtre, de sièges pour salles d'attente ou de chaises pour les établissements d'enseignement, se distinguent par un design moderne et ergonomique, ainsi que par une résistance à l'usure, une résistance et un confort élevés. Dans la vaste gamme de produits présentés dans le catalogue sur le site Web profkresla.ru, vous pouvez toujours choisir les modèles qui conviennent le mieux au style d'entreprise adopté dans votre entreprise. Si vous rencontrez toujours des difficultés avec le choix, les spécialistes de l'entreprise sont toujours prêts à vous conseiller, à vous aider à déterminer le modèle, puis à préparer un projet, à effectuer toutes les mesures et l'installation nécessaires sur place.
P En étudiant le sujet "Alcanes", nous montrons que les processus d'oxydation, de combustion, d'halogénation, de nitration, de déshydrogénation et de décomposition sont des processus redox. Lors de l'écriture des équations des réactions de combustion et de décomposition des substances organiques, il est préférable d'utiliser la valeur moyenne de s.d. carbone. Par exemple:
Nous prêtons attention à la première moitié de la balance électronique: à l'atome de carbone dans la valeur fractionnaire de s.d. le dénominateur est 4, nous calculons donc le transfert d'électrons à l'aide de ce coefficient.
Dans d'autres cas, lors de l'étude du sujet «Alcanes», nous déterminons les valeurs de s.d. chaque atome de carbone du composé, tout en attirant l'attention des élèves sur la séquence de substitution des atomes d'hydrogène au niveau des atomes de carbone primaires, secondaires et tertiaires :


Ainsi, nous amenons les étudiants à la conclusion qu'au début le processus de substitution a lieu au niveau du tertiaire, puis du secondaire et, en dernier lieu, au niveau des atomes de carbone primaires.
P Lors de l'étude du sujet "Alcènes", nous considérons les processus d'oxydation en fonction de la structure de l'alcène et du milieu réactionnel.
Lorsque les alcènes sont oxydés avec une solution concentrée de permanganate de potassium KMnO 4 en milieu acide (oxydation dure), les liaisons - et - se rompent avec formation d'acides carboxyliques, de cétones et de monoxyde de carbone (IV). Cette réaction est utilisée pour déterminer la position de la double liaison.
Si la double liaison se trouve à l'extrémité de la molécule (par exemple, dans le butène-1), l'un des produits d'oxydation est l'acide formique, qui s'oxyde facilement en dioxyde de carbone et en eau :

Nous soulignons que si dans la molécule d'alcène l'atome de carbone au niveau de la double liaison contient deux substituants carbone (par exemple, dans la molécule de 2-méthylbutène-2), alors lors de son oxydation une cétone se forme, puisque la transformation d'un tel atome dans un atome du groupe carboxyle est impossible sans casser la liaison C–C, relativement stable dans ces conditions :

Nous clarifions que si la molécule d'alcène est symétrique et que la double liaison est contenue au milieu de la molécule, alors un seul acide se forme lors de l'oxydation :

Nous signalons qu'une caractéristique de l'oxydation des alcènes, dans laquelle les atomes de carbone de la double liaison contiennent deux radicaux carbonés, est la formation de deux cétones :


Considérant l'oxydation des alcènes en milieu neutre ou faiblement alcalin, nous attirons l'attention des lycéens sur le fait que dans de telles conditions, l'oxydation s'accompagne de la formation de diols (alcools dihydriques), et des groupes hydroxyles sont attachés à ces atomes de carbone entre lesquels il y avait une double liaison :

DANS De manière similaire, on considère l'oxydation de l'acétylène et de ses homologues, selon le milieu dans lequel se déroule le processus. Ainsi, nous précisons que dans un environnement acide, le processus d'oxydation s'accompagne de la formation d'acides carboxyliques :

La réaction est utilisée pour déterminer la structure des alcynes par les produits d'oxydation :

En milieu neutre et légèrement alcalin, l'oxydation de l'acétylène s'accompagne de la formation des oxalates correspondants (sels de l'acide oxalique), et l'oxydation des homologues s'accompagne de la rupture de la triple liaison et de la formation de sels d'acides carboxyliques :

DANS Toutes les règles sont élaborées avec les étudiants sur des exemples précis, ce qui conduit à une meilleure assimilation de la matière théorique. Par conséquent, lors de l'étude de l'oxydation des arènes dans divers milieux, les élèves peuvent indépendamment faire des hypothèses selon lesquelles, dans un milieu acide, il faut s'attendre à la formation d'acides et, dans un milieu alcalin, de sels. L'enseignant n'aura qu'à préciser quels produits de réaction se forment en fonction de la structure de l'arène correspondante.
Nous montrons par des exemples que les homologues du benzène avec une chaîne latérale (quelle que soit sa longueur) sont oxydés par un agent oxydant fort en acide benzoïque au niveau de l'atome de carbone. Les homologues du benzène, lorsqu'ils sont chauffés, sont oxydés par le permanganate de potassium en milieu neutre pour former des sels de potassium d'acides aromatiques.
5C 6 H 5 -CH 3 + 6KMnO 4 + 9H 2 SO 4 \u003d 5C 6 H 5 COOH + 6MnSO 4 + 3K 2 SO 4 + 14H 2 O,
5C 6 H 5 -C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 \u003d 5C 6 H 5 COOH + 5CO 2 + 12MnSO 4 + 6K 2 SO 4 + 28H 2 O,
C 6 H 5 -CH 3 + 2KMnO 4 \u003d C 6 H 5 COOK + 2MnO 2 + KOH + H 2 O.
Nous soulignons que s'il y a plusieurs chaînes latérales dans une molécule d'arène, alors en milieu acide chacune d'entre elles est oxydée au niveau d'un atome de carbone a en un groupe carboxyle, entraînant la formation d'acides aromatiques polybasiques :

P Les compétences acquises dans la compilation des équations OVR pour les hydrocarbures permettent de les utiliser dans l'étude de la section « Composés oxygénés ».
Ainsi, lors de l'étude du sujet «Alcools», les élèves composent indépendamment les équations de l'oxydation des alcools, en utilisant les règles suivantes:
1) les alcools primaires sont oxydés en aldéhydes
3CH 3 -CH 2 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 \u003d 3CH 3 -CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O;
2) les alcools secondaires sont oxydés en cétones

3) pour les alcools tertiaires, la réaction d'oxydation n'est pas typique.
Afin de se préparer à UTILISER l'enseignant il convient de donner des informations complémentaires à ces propriétés, qui seront sans doute utiles aux étudiants.
Lorsque le méthanol est oxydé avec une solution acidifiée de permanganate de potassium ou de dichromate de potassium, du CO 2 se forme, des alcools primaires lors de l'oxydation, en fonction des conditions de réaction, peuvent former non seulement des aldéhydes, mais également des acides. Par exemple, l'oxydation de l'éthanol avec du bichromate de potassium à froid se termine par la formation d'acide acétique et, lorsqu'elle est chauffée, d'acétaldéhyde :
3CH 3 -CH 2 OH + 2K 2 Cr 2 O 7 + 8H 2 SO 4 \u003d 3CH 3 -COOH + 2K 2 SO 4 + 2Cr 2 (SO 4) 3 + 11H 2 O,
3CH 3 -CH 2 OH + K 2 Cr 2 O 7 + 4H 2 SO 4 3CH 3 -CHO + K 2 SO 4 + Cr 2 (SO 4) 3 + 7H 2 O.
Rappelons à nouveau aux étudiants l'influence de l'environnement sur les produits des réactions d'oxydation des alcools, à savoir : une solution neutre chaude de KMnO 4 oxyde le méthanol en carbonate de potassium, et les alcools restants en sels des acides carboxyliques correspondants :

Lors de l'étude du sujet "Aldéhydes et cétones", nous attirons l'attention des étudiants sur le fait que les aldéhydes sont plus facilement oxydés que les alcools en acides carboxyliques correspondants non seulement sous l'action d'agents oxydants forts (oxygène de l'air, solutions acidifiées de KMnO 4 et K 2 Cr 2 O 7), mais et sous l'influence de faibles (solution d'ammoniac d'oxyde d'argent ou d'hydroxyde de cuivre (II)):
5CH 3 -CHO + 2KMnO 4 + 3H 2 SO 4 \u003d 5CH 3 -COOH + 2MnSO 4 + K 2 SO 4 + 3H 2 O,
3CH 3 -CHO + K 2 Cr 2 O 7 + 4H 2 SO 4 \u003d 3CH 3 -COOH + Cr 2 (SO 4) 3 + K 2 SO 4 + 4H 2 O,
CH 3 -CHO + 2OH CH 3 -COONH 4 + 2Ag + 3NH 3 + H 2 O.
Nous portons une attention particulière à l'oxydation du méthanal avec une solution ammoniacale d'oxyde d'argent, car dans ce cas, il se forme du carbonate d'ammonium, et non de l'acide formique :
HCHO + 4OH \u003d (NH 4) 2 CO 3 + 4Ag + 6NH 3 + 2H 2 O.
Comme le montre notre expérience à long terme, la méthode proposée pour enseigner aux élèves du secondaire comment écrire des équations OVR avec la participation de substances organiques augmente leur finale UTILISER le résultat en chimie pour quelques points.